一种split-TEV-Fast系统的构建方法及其在检测蛋白相互作用中的应用与流程
本发明属于生物技术和分子生物学技术领域,尤其涉及一种split-tev-fast系统的构建方法及其在检测蛋白-蛋白相互作用中的应用。
背景技术:
研究蛋白-蛋白相互作用在诸多生物学领域中都具有极其重要的价值。在生物体内,一种蛋白质通常是与其他蛋白质发生相互作用来进一步执行其功能,是生物信息调控的主要方式,蛋白-蛋白相互作用阐明了整个细胞内的生理生化反应及分子机理,乃至细胞的整个生命过程。目前,验证蛋白-蛋白相互作用的方法主要包括几个经典的实验技术:酵母双杂交技术,免疫共沉淀技术(co-ip),gstpulldown技术等等。酵母双杂交技术的特点在于敏感、高效、操作简易可大规模的进行验证,在实际应用当中,由于酵母双杂交的敏感性而伴随着一定的假阳性,同时由于反应发生在细胞核内,所以也并非适用于所有的蛋白质,从而可应用co-ip,gstpulldown技术对初筛结果进行进一步的补充,但是co-ip使用免疫标签会有假阳性互作,gstpulldown需要体外表达纯化蛋白,也有一定的局限性。
本发明建立了一种全新的研究蛋白-蛋白相互作用的技术——yeastendoplasmicreticulumsequestrationbasedsplit-tev-fastsystem(yes-sts,基于酵母内质网滞留的split-tev-fast蛋白酶系统)。tevprotease(tobaccoetchvirusprotease)是来源于烟草蚀纹病毒(tev)的一种半胱氨酸蛋白酶,基于tev蛋白酶的强底物特异性以及高活性,新近建立起一种可用于研究蛋白间相互作用的split-tev蛋白酶系统,引用于:[wehrmc,laager,bolzu,fischertm,grunewalds,scheeks,etal.monitoringregulatedprotein-proteininteractionsusingsplittev.naturemethods.2006;3(12):985-93.]。split-tev以往所采用的均为野生型的tev蛋白酶,在待测目的蛋白的相互作用下重新形成完整的tev蛋白酶只保留其原有活性的40%,并且在细胞质中发挥作用,酶与底物作用空间大,浓度相对较低,造成反应灵敏度低,从而无法对蛋白互作强度进行准确定量。在本发明中,所用的tev-fast蛋白酶是经过蛋白酶工程、定向进化得到的一种高于野生型tev蛋白酶活性接近4倍的突变体,其活性更高,反应更加灵敏,引用于[yi,l.,gebhard,m.c.,li,q.,taft,j.m.,georgiou,g.,andiverson,b.l.“engineeringoftevproteasevariantsbyyeastersequestrationscreening(yess)ofcombinatoriallibraries”,proc.natl.acad.sci.u.s.a.,2013;110(18):7229-34]。如图1所示,tev-fast蛋白酶从118位被分拆成独立的-nh2端和-cooh端结构域,形成-nh2端(1-118)和-cooh端(119-242)tev-fast,-nh2端和-cooh端tev-fast结构域分别以接头(ggggsggggs或v2r)与待测目的蛋白相连,在待测目的蛋白的相互作用下重新形成完整的tev-fast蛋白酶,从而切割底物。由于野生型tev的活性较低,不足以使该系统发挥其灵敏性,故选用tev突变体tev-fast,保证该系统灵敏高效的特点,这是本发明的第一个创新点。
内质网滞留信号肽(ers,ersequestrationsignal)是蛋白质-cooh端的一段多肽,通常为4-6个氨基酸。大部分蛋白质在共翻译转运途径下进入内质网加工修饰,由于滞留信号肽与内质网内膜上的受体erd2的相互作用,可使得该蛋白质被滞留在内质网内一段时间。因为宿主细胞为真核酵母细胞,来源为真核细胞的目标蛋白能在其内更好的表达、折叠;并且内质网为大多数蛋白折叠、分泌必经场所,有大量的分子伴侣来有效维持蛋白稳定性,因此为研究蛋白互作提供了有力条件。此外,反应在内质网内发生,缩小了酶与底物反应的空间;而使用内质网滞留信号肽,可使酶与底物更长时间滞留在内质网,从而反应更充分,得以放大反应信号。应用不同强度的滞留信号肽可使得该split-tev-fast蛋白酶系统识别具有不同相互作用强度的互作蛋白,有效避免了酵母双杂得到的假阳性信号。同时,在不同强度ers的作用下,蛋白滞留在内质网腔中的时间也有所差异,运用目前已知强弱的滞留信号肽wehdel,fehdel,hdel及无滞留信号肽的对照组,可根据蛋白滞留在内质网内效应的强弱,酶与底物反应时间的长短,及最终检测的单荧光信号的比例来研究不同互作蛋白的相互作用强度,这是本发明的第二个创新点。
技术实现要素:
为了解决上述技术问题,本发明的目的之一在于提供一种新型的split-tev-fast系统,其中所使用的tev-fast酶的活性更强,在检测蛋白质之间的相互作用时检测灵敏度高。
为实现上述目的,本发明的技术方案是:一种split-tev-fast系统的构建方法,包括如下步骤:(1)构建载体pesd-ppi,所述载体pesd-ppi包含均受同一诱导剂诱导的一个正向启动子和一个双向启动子,所述正向启动子用以表达tev-fast蛋白酶的底物多肽和标签序列的对应的多肽,所述双向启动子用以表达拆分的tev-fast蛋白酶和待测的目的蛋白;(2)采用诱导剂诱导启动子,引发下游基因同时进行等量表达;(3)利用flag标签的荧光抗体和ha标签的荧光抗体对细胞进行荧光标记;(4)利用流式细胞仪检测细胞表面展示的单双荧光信号,再根据单双荧光信号的比例来判断蛋白与蛋白之间的相互作用的强度。
上述技术方案的有益效果在于:运用突变型的tev-fast酶,其酶活性接近野生型tev的四倍,大幅度提高整个系统的灵敏度;使用具有不同滞留强度的滞留信号肽,使蛋白酶对底物的切割发生在内质网中,并且停留不同的时间,产生不同的切割强度,再通过细胞表面的荧光信号被检测,因此可以用于蛋白相互作用的定量分析。
上述技术方案中所述标签序列为flag标签和ha标签。
上述技术方案中所述步骤(1)中正向启动子为表达aga2-flag-enlyfqs-ha-ers的gal1,所述双向启动子为gal1-gal10,分别用以表达拆分的-nh2端tev-fast蛋白酶-目的蛋白a和-cooh端tev-fast蛋白酶-目的蛋白b,并使拆分的tev-fast和目的蛋白之间用接头连接。其中所述ers为内质网滞留信号肽;所述接头为ggggsggggs或v2r,被人为拆分的-nh2和-cooh端tev-fast由于目的蛋白之间的相互作用,重构成完整的有活性的tev-fast以切割底物多肽序列enlyfq↓s,形成aga2-flag-enlyfq,显示单荧光信号;如果不能重构成完整的有活性的tev-fast,底物不被切割,形成aga2-flag-enlyfqs-ha-ers,显示双荧光信号。
上述技术方案的有益效果在于:在pesd-ppi的质粒内采用1个正向的启动子gal1表达底物多肽enlyfqs与标签序列和1个双向启动子gal1-gal10表达拆分的酶和待测的互作蛋白,其中双向启动子的表达量近似为1:1,可保证拆分的tev-fast的两部分同时表达且表达量一致,在被拆分的tev-fast蛋白酶的n端加上一段内质网定位信号肽,使其可进入内质网内,在酵母细胞中,在诱导剂的作用下,酵母菌内的转录激活蛋白与gal启动子的上游激活序列结合并使得胞内同时表达底物多肽序列及tev-fast蛋白酶和目的蛋白,基于tev-fast使该系统得以实现,再在拆分的-cooh端和-nh2端tev-fast蛋白酶与目的蛋白的融合体的cooh端都分别加上不同强度的滞留信号肽,分泌性蛋白经过内质网时由于内质网滞留信号肽与erd2(内质网滞留信号肽受体蛋白)的作用,滞留在内质网内,使得反应在内质网中进行,并且滞留信号肽越强,在内质网滞留的时间越长。
上述技术方案中所述诱导剂为半乳糖,可同时诱导所述正向启动子和所述双向启动子等量表达。
上述技术方案的有益效果在于:其中,酵母菌的启动子gal1、gal1-gal10受半乳糖调控,使正向启动子和双向启动子同时表达下游基因,从而使得整个反应便于控制。
上述技术方案中所述aga2为酿酒酵母表面的α凝集素的结合亚单位,可与酵母表面的膜蛋白aga1以二硫键结合展示于酵母表面,其中aga1为α凝集素核心亚单位,在aga2蛋白的牵引下,便可将底物多肽序列展示于细胞表面。
上述技术方案的有益效果在于:使得胞内表达的产物能够被分泌至胞外,并被展示在细胞的表面,便于检测荧光信号。
本发明的目的之二在于将如上所述的split-tev-fast系统应用在检测蛋白相互作用中,其检测灵敏度高,且可对互作蛋白作用强度进行定量,检测具有不同互作强度的蛋白。
该系统的基础是构建含有split-tev-fast、目的基因和tev-fast蛋白酶底物、荧光标签序列的载体,并转化于eby200(matagal1-aga1::ura3kex2δura3-52trp1leu2δ1his3δ200pep4:his3prb1δ1.6rcan1gal)酵母细胞,且该系统结合了内质网滞留信号肽的特点与功能,成立一套新的系统。该系统进行在酵母体内内质网,且信号展示在细胞表面,从而得以检测。并且单双荧光信号是由流式细胞仪检测到的,且单双信号各代表一种生物学意义,即蛋白质相互作用或蛋白质不相互作用。
附图说明
图1为split-tev-fast系统检测蛋白相互作用的原理图;
图2为载体pesd-ppi的质粒图谱;
图3为目的蛋白分别为yae1与lto1的流式细胞仪检测图;
图4为目的蛋白分别为rli1与lto1的流式细胞仪检测图;
图5为目的蛋白为yae1和lto1,滞留信号肽为wehdel时流式细胞仪检测到的结果;
图6为目的蛋白为yae1和lto1,滞留信号肽为fehdel时流式细胞仪检测到的结果;
图7为目的蛋白为yae1和lto1,信号肽为hdel时流式细胞仪检测到的结果;
图8为目的蛋白为yae1和lto1,无滞留信号肽时流式细胞仪检测到的结果;
图9为目的蛋白为rli1和lto1时,滞留信号肽为wehdel时使用流式细胞仪检测到的结果;
图10为目的蛋白为rli1和lto1时,滞留信号肽为fehdel时使用流式细胞仪检测到的结果;
图11为目的蛋白为rli1和lto1时,滞留信号肽为hdel时使用流式细胞仪检测到的结果;
图12为目的蛋白为rli1和lto1时,无滞留信号肽时使用流式细胞仪检测到的结果。
具体实施方式
以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
实施例1
含有滞留信号肽wehdel重组载体构建过程
(1)首先通过聚合酶链式反应(pcr)扩增得到aga2-flag-enlyfqs-ha-ers(wehdel)目的基因,反应体系如下:50μl体系,10×kodbuffer,5μl;dntp(2.5mm),3μl;引物f1(10μm),2μl;引物r1(10μm),2μl;pfu聚合酶,1μl;template(含tev蛋白酶底物多肽序列),0.5μl(20ng/μl);加双蒸水至50μl。
pcr扩增条件:95℃,5min;95℃,30s,57℃,30s,72℃,30s,25个循环;72℃,5min;4℃,∞。
引物设计如下:
f1为seqidno.1
r1为seqidno.2
(2)琼脂糖凝胶回收得到目的片段,分别用xhoi,ecori37℃双酶切6hr,再用琼脂糖凝胶回收,双酶切体系:目的片段dna,30μl;buffercutsmart,5μl;xhoi,ecori,各2μl;加双蒸水至50μl。
(3)以同样的体系酶切载体pesd并回收,16℃酶连载体和目的基因18hr,酶连体系:酶切载体,1.2μl;酶切目的基因,0.5μl;t4dnaligase,0.2μl(thermofisher),t4ligasebuffer,2μl;加双蒸水至20μl。
(4)转化。取5μl酶连产物于50μlxl-glod感受态中,冰上静置10min后42℃热激35s,加lb培养基于37℃,250rpm摇床内培养45-60min后,涂布amp抗性平板。
(5)各挑2个经过菌落pcr鉴定的重组子测序,得到正确重组的表达底物载体。
(6)在该载体基础上继续构建-nh2端tev-fast部分,首先用引物wtp5、wtp6扩增获得-nh2端tev-fast,再以此片段为模板用引物wtp5及反向引物wtp7、wtp8扩增以带上接头序列v2r与目的基因ayae1同源的20bpdna序列。以引物wtp9及反向引物wtp10、wtp11扩增获得携带内质网定位信号肽的目的基因ayae1,再通过overlapingpcr的方法用引物wtp5和wtp11将-nh2端tev-fast与yae1连接成完整片段,且该完整片段的-cooh端已加上滞留信号肽wedhel,琼脂糖凝胶回收该片段。扩增引物如下:
wtp5为seqidno.3;
wtp6为seqidno.4;
wtp7为seqidno.5;
wtp8为seqidno.6;
wtp9为seqidno.7;
wtp10为seqidno.8;
wtp11为seqidno.9;
(7)双酶切步骤(5)(6)得到的重组载体和完整目的片段6hr,双酶切体系:目的片段dna,35μl;buffercutsmart,5μl;bamhi,psti,各2μl;加双蒸水至50μl。琼脂糖凝胶回收载体和片段,再以t4dnaligase酶连,转化于xl-gold,待长出菌落后挑选重组子进行测序。
(8)在该基础上继续构建-cooh端tev-fast部分,首先用正向引物wtp13和反向引物wtp14、wtp22分别扩增目的基因blto1和rli1,并用引物wtp15带上接头序列ggggsggggs和与-cooh端tev-fast同源的20bpdna序列,再用引物wtp16、wtp17扩增得到-cooh端tev-fast,以overlapingpcr的方法用引物wtp13、wtp17将-cooh端tev-fast和两个目的基因b连接成完整片段,且该完整片段的-cooh端已加上滞留信号肽wedhel,琼脂糖凝胶回收该片段。选用酶切位点sali,ndei,如上述方法酶切酶连转化于xl-gold,得到最终的载体后可进行进一步的验证。扩增引物如下:
wtp13为seqidno.10;
wtp14为seqidno.11;
wtp15为seqidno.12;
wtp16为seqidno.13;
wtp17为seqidno.14;
wtp22为seqidno.15;
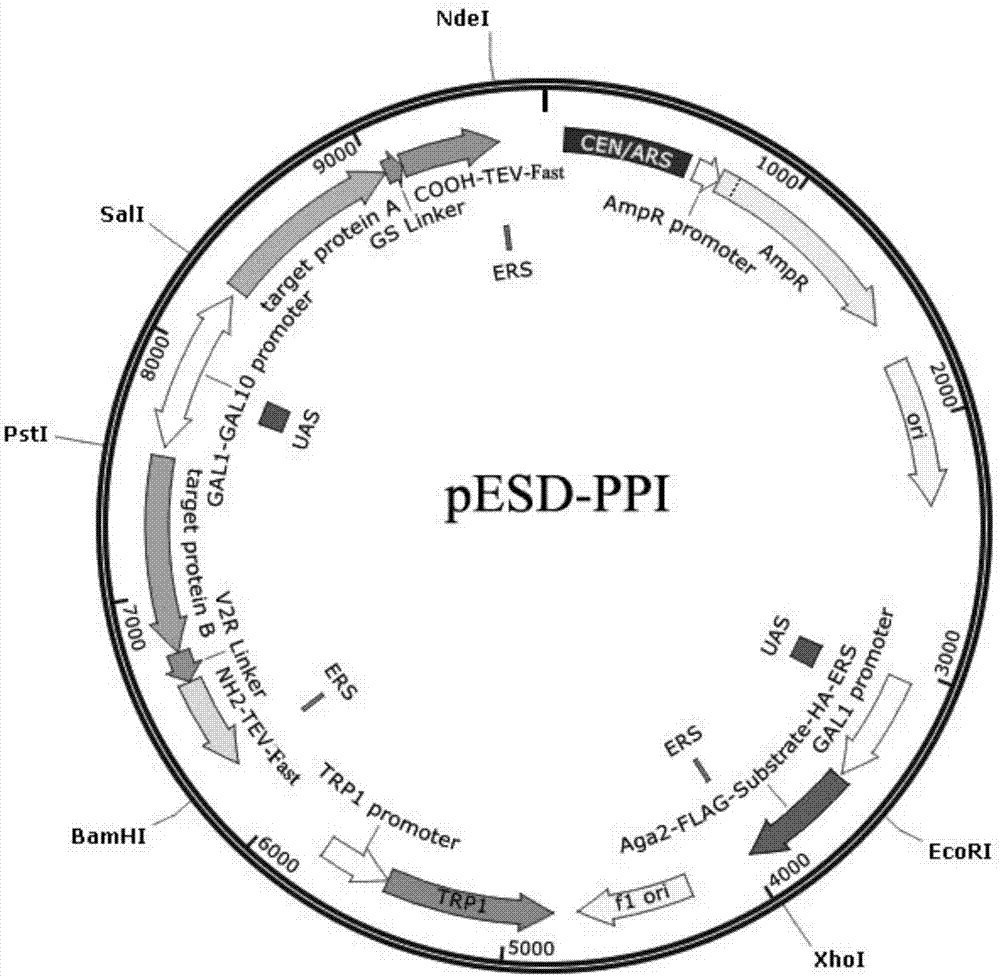
最终构建得到载体pesd-ppi(图2)注释:
底物端:gal1-aga2-flag-enlyfqs-ha-ers(wehdel)、
tev-fast与目的蛋白端:gal10-yae1-nh2-tev-fast-ers(wehdel)或gal10-rli1-nh2-tev-fast-ers(wehdel)、gal1-lto1-tev-fast-cooh-ers(wehdel)。
实施例2
含有split-tev-fast系统的重组载体的诱导表达
将最终载体转化于酿酒酵母eby200转化所用的试剂为:eby200感受态,20μl;重组质粒,1.5μl;singlestrandcarrierdna,25μl;1mliac,36μl;peg4000(50%w/v),240μl;混合体系后,30℃水浴30min,转移至42℃热激20min,离心收集菌体,加1mlypd液体培养基于30℃摇床培养1.5hr,用超纯水洗细胞一次后,涂布于ynb-caa-glucose固体培养基,30℃培养箱培养2-3days。ynb-caa-glucose固体培养基成分为20g/l葡萄糖,6.7g/lynb,5.4g/lna2hpo4,8.6g/lnah2po4·h2o,5g/lcasaminoacids,ph7.4,15g/l琼脂。
接种单菌落于液体培养基ynb-caa-glucose中,30℃,230rpm摇床中培养至od600=3左右,转移至ynb-caa-galactose液体培养基于30℃、230rpm摇床中培养,并使起始od600=0.8,诱导8hr后取样,ynb-caa-galactose液体培养基成分中用半乳糖替代葡萄糖,起诱导表达的作用。
实施例3
应用流式细胞仪检测split-tev-fast系统检测蛋白相互作用的结果
选用一个由yae1,lto1,rli1组成的复合体来验证split-tev-fast系统,其中lto1和yae1之间具有相互作用,与rli1之间不具有相互作用。于是将yae1和lto1设为实验组,rli1和lto1为负对照组,由于yae1和lto1之间的相互作用,促使-nh2端和-cooh端split-tev-fast重构成完整的有活性的tev-fast以切割底物,酵母表面则展示aga2-flag-enlyfq,随后通过anti-flag-ifluor647荧光抗体标记检测到单荧光信号ifluor647。而当目的蛋白为rli1和lto1时,在二者不发生相互作用的情况下,则不能形成完整的有活性的tev-fast蛋白酶,无法切割-enlyfqs,酵母表面则展示aga2-flag-enlyfqs-ha,随后通过anti-flag-ifluor647和anti-ha-fitc荧光抗体标记检测到双荧光信号(ifluor647和fitc),说明该系统可以快速方便的检测到蛋白之间的相互作用。标记酵母表面时所使用到的试剂有anti-flag-ifluor647和anti-ha-fitc抗体,以及buffera(1xpbs+0.5%w/vbsa+1mmedtaph7.4)、bufferb(1xpbs+0.5%w/vbsaph7.4)、bufferc(1xpbsph7.4)以洗去细胞表面的杂质。
具体操作步骤如下,取106个细胞,加150μlbuffera液混匀后离心沉淀细胞,3000rpm,4℃,1min。再用bufferb液洗细胞,去上清,然后每个样品加20μlbufferb液,0.15μlfitc,0.15μlifluor647,4℃避光标记15min,转移至室温避光标记30min。离心去上清,再用150μlbufferb洗一次,继续用1xpbs洗一次后,并用300μl重悬。重悬的细胞用于cytoflex流式细胞仪分析,检测的荧光信号通道是fitc(bp525/40nm)和apc(bp660/20nm)。从流式细胞仪中给出的结果分析得出该系统可以灵敏的检测到yae1和lto1蛋白之间的相互作用,并能确定目的蛋白yae1和rli1间的非相互作用。
具体如图3-4所示,横坐标指示的是anti-flag-ifluor647荧光信号强度,纵坐标显示的是anti-ha-fitc荧光信号强度,从流式细胞仪检测到的结果分析,图3显示的是yae1和lto1的流式细胞图,anti-flag-apc荧光信号占总展示比例的95%,几乎只检测到anti-flag-apc荧光,说明待测蛋白之间的相互作用使得形成完整的tev-fast蛋白酶,在内质网内进行反应切割底物,故只有flag标签被展示到酵母表面。当待测蛋白rli1和lto1间不相互作用,流式细胞仪检测到双荧光信号,目的蛋白之间不相互作用,未能形成完整的tev-fast,底物不被切割。
实施例4
利用不同强度滞留信号肽调整split-tev-fast系统以检测蛋白与蛋白相互作用强度
在实施例1构建的含有滞留信号肽wehdel重组载体的基础上,分别用fehdel,hdel及无滞留信号肽替代wehdel,构建含有fehdel,hdel及无滞留信号肽的重组载体,分别转化酵母细胞,诱导表达,荧光抗体标记后利用cytoflex流式细胞仪检测结果。
引物设计如下:
1、扩增底物多肽序列(反向引物中包含fehdel,hdel及无滞留信号肽)
正向引物11f为seqidno.16;
反向引物11r为seqidno.17;
反向引物12r为seqidno.18;
反向引物13r为seqidno.19;
2、扩增nh2-tev-targetprotein(正向引物中包含fehdel,hdel及无滞留信号肽)
正向引物12f为seqidno.20;
正向引物13f为seqidno.21;
正向引物14f为seqidno.22;
反向引物14r为seqidno.23;
2、扩增cooh-tev-targetprotein(反向引物中包含fehdel,hdel及无滞留信号肽)
正向引物15f为seqidno.24;
反向引物15r为seqidno.25;
反向引物16r为seqidno.26;
反向引物17r为seqidno.27;
将构建好的分别含有wehdel,fehdel,hdel及无滞留信号肽的系列重组载体按实施例3中所述方法分别转化酿酒酵母感受态eby200中,以实施例3中所给方法进行诱导表达,荧光标记后使用cytoflex流式细胞仪检测,结果显示当使用不同强度的内质网滞留信号肽(ers)时,酵母表面展示出一定梯度比例的单荧光信号,如图5-8所示,为目的蛋白分别为yae1和lto1时相互作用的效果,当使用的滞留信号肽(ers)强度逐渐减弱时(wehdel>fehdel>hdel>none),单荧光信号占总荧光信号的比例逐渐降低,比例为95%>70%>20%>0。由于在不同内质网滞留效果的情况下,可检测到的表示相互作用的单荧光信号所占比例有明显差异,滞留效果越强,在内质网内tev-fast蛋白酶与底物反应的时间越长,单荧光信号比例越高,当滞留效果逐渐减弱,单信号也逐渐降低。由此可确定,本split-tev-fast系统具有明显的动力学范围,可以运用不同强度的滞留信号肽去检测蛋白与蛋白间不同强度的相互作用。而图9-12为不能相互作用的两个蛋白rli1和lto1的实验结果,如图9-12所示,不管用强滞留信号肽还是没有滞留信号肽,tev-fast蛋白酶都不能重构,不能切割底物,表现为双荧光信号,充分说明本方法的严谨性。
该系统相较于酵母双杂交系统,可以有效的减少假阳性,因为酵母双杂交发生在细胞核内,通过报告基因的表达与否来确定是否具有相互作用。由于核定位而不可避免的造成假阳性的产生,而在该系统中,反应定位于内质网内,可以有效避免假阳性,并且通过不同的内质网滞留信号肽可以测相互作用的强弱。
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
序列表
<110>湖北大学
<120>一种split-tev-fast系统的构建方法及其在检测蛋白相互作用中的应用
<160>27
<170>siposequencelisting1.0
<210>1
<211>31
<212>dna
<213>人工序列(artificialsequence)
<400>1
cggaattcatgcagttacttcgctgtttttc31
<210>2
<211>51
<212>dna
<213>人工序列(artificialsequence)
<400>2
ccgctcgagtcacaattcgtcgtgttcccatgagccagcgtagtctggaac51
<210>3
<211>56
<212>dna
<213>人工序列(artificialsequence)
<400>3
cgggatcctcacaattcgtcgtgttcccatgagccagtttggaagttggttgtcac56
<210>4
<211>56
<212>dna
<213>人工序列(artificialsequence)
<400>4
gcgagcccgagcaacccgggcgcgagcaacggcagcggagaaagcttgtttaaggg56
<210>5
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>5
ccgcgagcagcagcctggcgaaagataccagcagcgcgagcccgagcaacccggg55
<210>6
<211>55
<212>dna
<213>人工序列(artificialsequence)
<400>6
gccgagcctgggcccgcaggatgaaagctgcaccaccgcgagcagcagcctggcg55
<210>7
<211>56
<212>dna
<213>人工序列(artificialsequence)
<400>7
cctgcgggcccaggctcggcggggtgcggccgcgcgcttggatagatggcacatgg56
<210>8
<211>51
<212>dna
<213>人工序列(artificialsequence)
<400>8
caatattttctgttattgcttcagttttagcaatgtcgaatacttgggacg51
<210>9
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>9
gcactgcagatgcagttacttcgctgtttttcaatattttctgttattgc50
<210>10
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>10
gcgtcgacatgcaacttttgagatgcttcagtattttcagcgtcatcgcc50
<210>11
<211>49
<212>dna
<213>人工序列(artificialsequence)
<400>11
gctgccgccgccgccgctgccgccgccgccaataccggtgttatccaag49
<210>12
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>12
caccatgctagacatgctcttgctgccgccgccgccgctgc41
<210>13
<211>33
<212>dna
<213>人工序列(artificialsequence)
<400>13
caagagcatgtctagcatggtgtcagacactag33
<210>14
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>14
cgccatatgtcacaattcgtcgtgttcccatgagccctggctatacaccagttcattc58
<210>15
<211>51
<212>dna
<213>人工序列(artificialsequence)
<400>15
gctgccgccgccgccgctgccgccgccgccccaggattgagcctgattttg51
<210>16
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>16
cggaattcatgcagttacttcgctgttttt30
<210>17
<211>36
<212>dna
<213>人工序列(artificialsequence)
<400>17
ccgctcgagtcacaattcgtcgtgttcgaatgagcc36
<210>18
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>18
ccgctcgagtcacaattcgtcgtgtgagcc30
<210>19
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>19
ccgctcgagtcatgagcc18
<210>20
<211>37
<212>dna
<213>人工序列(artificialsequence)
<400>20
cgggatcctcacaattcgtcgtgttcgaatgagccag37
<210>21
<211>31
<212>dna
<213>人工序列(artificialsequence)
<400>21
cgggatcctcacaattcgtcgtgtgagccag31
<210>22
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>22
cgggatcctcatgagccag19
<210>23
<211>26
<212>dna
<213>人工序列(artificialsequence)
<400>23
gcactgcagatgcagttacttcgctg26
<210>24
<211>29
<212>dna
<213>人工序列(artificialsequence)
<400>24
gcgtcgacatgcaacttttgagatgcttc29
<210>25
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>25
agggctcattcgaacacgacgaattgtgacatatggcg38
<210>26
<211>32
<212>dna
<213>人工序列(artificialsequence)
<400>26
agggctcacacgacgaattgtgacatatggcg32
<210>27
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>27
agggctcatgacatatggcg20
- 还没有人留言评论。精彩留言会获得点赞!