一种树枝状四聚阳离子季铵盐型表面活性单体的制备方法与流程
本发明涉及一种表面活性单体的制备方法,具体涉及一种季铵盐阳离子表面活性单体的制备方法,特别涉及一种同时含有树枝状结构和四聚体阳离子的季铵盐型表面活性单体的制备方法。
背景技术:
树枝状四聚阳离子季铵盐型表面活性剂同时具有树枝状结构和碳氢疏水基团,具有较高的表面活性、乳化稳定性和聚集性能,可形成多种结构的聚集体,如类似人脑的囊泡、纳米聚集体及多相分隔胶束,可被应用于如药物载体、石油降黏及污水净化处理等诸多领域,成为目前研究的热点之一。表面活性单体,或称可聚合表面活性剂(surfmer),是一种功能性表面活性剂,这种表面活性剂分子不仅包含亲水头基、疏水尾基和可聚合的双键基团,而且还有独特的树枝状结构和四聚阳离子体。这种特殊的分子结构赋予它们独特的物理化学性质:(1)与普通表面活性剂相似,它们具有表面活性;(2)与乙烯类单体相似,它们能够被引发剂引发发生聚合反应;(3)树枝状结构可以使它的亲油部分更好地与油溶性物质融合,从而更好地形成乳化作用;(4)四聚阳离子体可以使它的阳离子度更大,疏水缔合性能、电荷中和能力和表面活性都优于一般的表面活性剂。表面活性单体的两亲性和可聚合性使它们在很多科技领域应用广泛。例如,可聚合表面活性单体可以用于石油开采行业对于稠油的乳化降粘;可以用于对油田的大量含油废水进行破乳处理;还可以作为可聚合的乳化剂改善乳液聚合反应。树枝状四聚阳离子季铵盐型表面活性单体是一种引入长链树枝状疏水基团的季铵盐型表面活性单体。与一般的阳离子表面活性单体相比,树枝状表面活性单体具有高表面活性,高耐热稳定性和高化学稳定性等优异的物理化学性能;将其与丙烯酰胺共聚得到的聚丙烯酰胺疏水缔合衍生物表现出良好的增稠效果和耐温抗盐能力。
目前,国内许多研究人员对可聚合表面活性单体的制备方法进行了研究,通常合成方法复杂,产物纯化困难,产物收率低,单体合成之后表面活性较低,后期用于聚合时还需要另外加入乳化剂,以促进油溶性单体和水溶性单体均相聚合,但是乳化剂的加入不但使得终产物无法提纯干净,而且乳化剂这类小分子在聚合过程中也会促进链转移,使得聚合反应的分子量偏低,从而影响聚合物的最终性能。另外,关于树枝状四聚阳离子季铵盐型表面活性单体的合成国内尚未见公开研究报道。
技术实现要素:
鉴于现有技术中存在的可聚合表面活性单体的制备方法复杂,产物纯化困难,产物收率低,单体合成之后表面活性较低,后期用于聚合时还需要另外加入乳化剂,使得聚合反应的分子量偏低的技术问题,本发明提供了一种树枝状四聚阳离子季铵盐型表面活性单体的制备方法。
为了实现上述目的,本发明采用如下技术方案:
一种树枝状四聚阳离子季铵盐型表面活性单体的制备方法,反应过程如下反应式所示:
包括如下步骤:
步骤(i),将丙烯酸甲酯和单体3,3-二氨基-n-甲基二丙胺充分混合进行加成反应,使单体3,3-二氨基-n-甲基二丙胺两端的伯胺基团转化为酯基,得到含所述化合物ⅰ的溶液;
步骤(ii),加入6-溴-1-己烯与所述含所述化合物ⅰ的溶液进行季铵化反应,反应结束后,减压除去溶剂,然后加入无水乙醚作为沉淀剂洗涤三次,得到所述化合物ⅱ;
步骤(iii),加入n,n-二甲基-1,3-丙二胺与所述化合物ⅱ进行酰胺化反应,反应结束后,减压除去过量的n,n-二甲基-1,3-丙二胺,得到化合物ⅲ;
步骤(iv),加入长链单体1-溴十二烷与所述化合物ⅲ进行季胺化反应,反应结束后反复洗涤萃取三次以上,得到纯净的化合物ⅳ;
所述化合物ⅳ为所述树枝状四聚阳离子季铵盐型表面活性单体。
本发明的有益效果包括:原料易得,合成步骤及产物纯化简单,收率高,使用安全,储存方便,适于工业化生产,并且具有广泛的应用前景。
所合成的表面活性单体是一种树枝状四聚阳离子季铵盐型表面活性剂,应用广泛,如制成石油乳化降黏剂、织物柔软剂、杀菌剂、药物载体、废水处理絮凝剂等,可与多种单体通过无皂乳液聚合制成各种功能性大分子,可广泛应用于油田开发、涂布、消防、印刷、电镀、日用化工、医药等领域。
进一步,步骤(i)所述3,3-二氨基-n-甲基二丙胺和丙烯酸甲酯的摩尔比为1:6~8,溶剂为甲醇或乙醇;3,3-二氨基-n-甲基二丙胺与所述溶剂的质量分数比为1:10~16,反应温度为25~40℃,反应时间为24~36小时,投料方式是将丙烯酸甲酯溶于所述溶剂中,在黑暗条件下,于0.5~1小时内逐滴滴入3,3-二氨基-n-甲基二丙胺中,所述反应在氮气氛围下进行。
采用上述进一步的有益效果是:利用迈克尔加成法合成化合物i,此法副产物极少,且产物后处理简单,纯度高,收率高。由于用丙烯酸甲酯对氨基进行的迈克尔加成反应一般都极易进行,且丙烯酸甲酯相对3,3-二氨基-n-甲基二丙胺过量,故可保证单体3,3-二氨基-n-甲基二丙胺反应完全。接上丙烯酸甲酯之后,为表面活性剂的整体树枝状结构奠定了基础,利于通过之后的酰胺化反应对端基进行改性,过量的丙烯酸甲酯沸点低,很容易通过减压的方法除去,所以后期产物纯化简单。
进一步,步骤(ii)所述含所述化合物ⅰ的溶液和6-溴-1-己烯的摩尔比为1:1.3~1.5,溶剂为甲醇、乙醇或丙酮,所述化合物i与所述溶剂的质量分数比为3:3~5,所述6-溴-1-己烯与所述溶剂的质量分数比为1.5:2~4,反应温度为25~30℃,反应时间为72~96小时,投料方式是将6-溴-1-己烯溶于所述溶剂中,然后在黑暗条件下,于0.5~1小时内逐滴滴入所述含所述化合物ⅰ的溶液的中,反应在氮气氛围下进行。
采用上述进一步的有益效果是:生成化合物ⅱ的季胺化反应操作简单方便,使分子被引入可用于聚合的双键,且产物后处理简单,纯度高,收率高。由于合成的化合物ⅱ通过季胺化反应引入双键,具备单个季铵盐的结构,利用其和反应物之间极性的差异,使用洗涤剂乙醚很容易可以分离提纯出纯净的产物。
进一步,步骤(iii)所述反应溶剂为甲醇,所述化合物ⅱ和n,n-二甲基-1,3-丙二胺的摩尔比为1:25~40,所述化合物ⅱ与溶剂的质量分数比为1:10~16,温度为25~30℃,反应时间为72~96小时,投料方式是将所述化合物ⅱ溶于所述反应溶剂中,并在黑暗条件下,在0.5~1小时内逐滴滴入n,n-二甲基-1,3-丙二胺中;所述n,n-二甲基-1,3-丙二胺在反应之置于冰水浴中,所述步骤(iii)在氮气氛围下进行。
采用上述进一步的有益效果是:化合物iii合成简单,产物收集方便,纯度高。通过丙烯酸甲酯末端的酯基和n,n-二甲基-1,3-丙二胺之间的酰胺化反应,轻松引入用于最后一步反应所需要的叔胺。此种合成方法操作简单,反应条件温和、易控制,毒性低,收率高。
进一步,步骤(iv)所述化合物ⅲ和1-溴十二烷的摩尔比为1:6~8,反应溶剂为丙酮,所述化合物iii与所述反应溶剂的质量分数比为3:8~10,所述1-溴十二烷与所述反应溶剂的质量分数比为6:5~7,反应温度为30~40℃,反应时间为72~96小时,投料方式为将化合物ⅲ溶于所述反应溶剂中,于0.5~1小时内逐滴滴入过量的1-溴十二烷中,反应在氮气氛围下进行;所述洗涤萃取为以石油醚反复洗涤萃取。
采用上述进一步的有益效果是:操作简单,纯化方便,产物收率高。由于1-溴十二烷相对化合物ⅲ过量1.5~2倍,故可保证化合物ⅲ反应完全;而且生成的终产物化合物ⅳ的极性较大,所用反应物1-溴十二烷的极性很小,利用它们在石油醚中溶解度的差异,即可分离出纯净的产物iv。
本发明有益效果还包括:整个合成过程所需温度较低,大部分步骤室温就可完成,操作简单易控制,能耗低,适于工业大规模合成。
附图说明
图1为本发明树枝状四聚阳离子季铵盐型表面活性单体合成路线示意图;
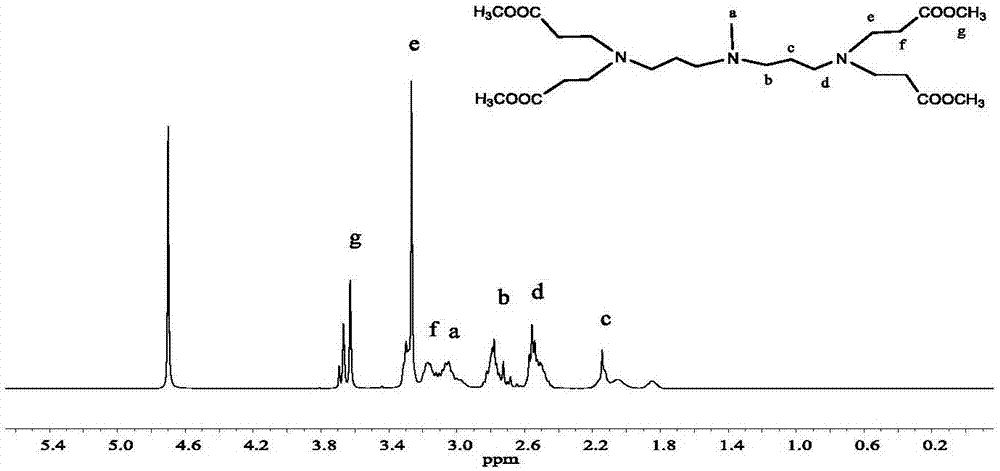
图2为本发明实施例1制备的化合物i的1h-nmr谱图;
图3为本发明实施例1制备的化合物ii的1h-nmr谱图;
图4为本发明实施例1制备的化合物iii的1h-nmr谱图;
图5为本发明实施例1制备的化合物iv的1h-nmr谱图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围
下列实施例中所使用的实验方法如无特殊说明,均为常规方法。
以下试剂均从商业途径得到,未做进一步纯化处理:3,3-二氨基-n-甲基二丙胺(98%,天津希恩思生化科技有限公司),丙烯酸甲酯(99%,北京百灵威科技有限公司),6-溴-1-己烯(98%,天津法莫西科技有限公司),n,n-二甲基-1,3-丙二胺(99%,上海阿拉丁生化科技股份有限公司),1-溴十二烷(98%,北京百灵威科技有限公司)。
以下试剂从商业途径得到,并作进一步纯化处理:甲醇、无水乙醚、丙酮购自烟台市双双化工有限公司,均为分析纯,参照purificationoflaboratorychemicals,5thed(chai,c.l.l.;armarego,w.l.f.,butterwirth-heinemann:newyork,2003.)处理后使用。
实施例1
(1)化合物i的制备
搭建反应装置,包括水浴锅,三口烧瓶,球形冷凝管,干燥管,氮气供给装置。首先将搭建好的装置置于冰水浴中,并通氮气5min,取2.9g(20mmol)3,3-二氨基-n-甲基二丙胺并溶于5ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶内,再通氮气20min,然后量取13.76g(160mmol)丙烯酸甲酯在黑暗条件下以滴/3s的速度加入三口瓶中,滴加结束后待体系自然恢复至室温,然后设定30℃,反应24h。反应结束后,旋蒸除去甲醇,并反复加入无水甲醇旋蒸除去丙烯酸甲酯,循环5次以上,最终得到8.65g淡黄色微粘液体即化合物ⅰ,收率为88.4%,由化合物ⅰ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)2.15(4h,m,-ch2(ch3)-n-[ch2-ch2-ch2]2-),2.57(4h,t,-ch2(ch3)-n-[ch2-ch2-ch2]2-),2.8(4h,t,-ch2(ch3)-n-[ch2-ch2-ch2]2-),3.04(3h,s,-ch2(ch3)-n-[ch2-ch2-ch2]2-),3.18(8h,t,[cooch3-ch2-ch2]4-n),3.28(8h,t,[cooch3-ch2-ch2]4-n),3.62(12h,s,[cooch3-ch2-ch2]4-n)。
(2)化合物ii的制备
装置通氮气5min,然后取2.934g(6mmol)化合物ⅰ溶于4ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶内,再通氮气20min,取1.467g(9mmol)6-溴-1己烯溶于3ml无水甲醇中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加结束后体系在30℃下反应72h。反应结束后,旋蒸除去甲醇,并用无水乙醚反复萃取提纯,最终得到3.35g黄色黏性液体即化合物ⅱ,收率为85.6%,由化合物ⅱ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)1.48(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.68(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.86(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.10(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.51(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.75(8h,t,[cooch3-ch2-ch2]4-n),3.0(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.25(3h,s,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.3(8h,t,[cooch3-ch2-ch2]4-n),3.66(12h,s,[cooch3-ch2-ch2]4-n),3.67(2h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),5.05(2h,t,ch2-ch=ch2),5.84(1h,m,ch2-ch=ch2)。
(3)化合物iii的制备
将装置置于冰水浴中,通氮气5min,然后取20.4g(200mmol)n,n-二甲基-1,3-丙二胺溶于15ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶中,在黑暗条件下搅拌并通氮气20min,取3.26g(5mmol)化合物ⅱ溶于5ml无水甲醇中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加完成后待体系自然恢复至室温,再设定温度30℃,黑暗条件下反应96h。旋蒸除去甲醇,并反复加入甲醇旋蒸除去过量的n,n二甲基1,3丙二胺,循环多次处理之后,最终得到4.19g淡黄色粘稠液体即化合物ⅲ,收率为90%,由化合物ⅲ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)1.41(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.62(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.82(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.15(24h,s,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.16(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.34(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.49(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.72(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.96(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),3.11(6h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.2(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),3.26(3h,s,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),5.0(2h,t,ch2-ch=ch2),5.8(1h,m,ch2-ch=ch2)。
(4)化合物iv的制备
装置通氮气5min,然后取5.98g(24mmol)1-溴十二烷溶于6ml无水丙酮中,加入上接球形冷凝管和干燥管的三口烧瓶中,再通氮气20min,取2.80g(3mmol)化合物ⅲ溶于9ml无水丙酮中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加完成设置30℃,反应72h。反应结束后,旋蒸除去丙酮,并用石油醚反复洗涤萃取,并用真空烘箱烘干,最终得到4.92g桔黄色颗粒状固体即化合物ⅳ,收率为85%,由化合物ⅳ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)0.82(12h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),1.21(88h,m,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),1.3(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.42(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.7(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),1.96(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.4(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.57(8h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),3.08(27h,s,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.25(14h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2,-ch2(ch3)n+-[ch2-ch2-ch2-n]2,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),3.35(8h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),5.0(2h,t,ch2-ch=ch2),5.8(1h,m,ch2-ch=ch2)。
实施例2
(1)化合物i的制备
搭建反应装置,包括水浴锅,三口烧瓶,球形冷凝管,干燥管,氮气供给装置。首先将搭建好的装置置于冰水浴中,并通氮气5min,取2.175g(15mmol)3,3-二氨基-n-甲基二丙胺并溶于10ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶内,再通氮气20min,然后量取8.6g(100mmol)丙烯酸甲酯在黑暗条件下以滴/3s的速度加入三口瓶中,滴加结束后待体系自然恢复至室温,然后设定25℃,反应30h。反应结束后,旋蒸除去甲醇,并反复加入无水甲醇旋蒸除去丙烯酸甲酯,循环5次以上,最终得到6.10g淡黄色微粘液体即化合物ⅰ,收率为83.2%,由化合物ⅰ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)2.15(4h,m,-ch2(ch3)-n-[ch2-ch2-ch2]2-),2.57(4h,t,-ch2(ch3)-n-[ch2-ch2-ch2]2-),2.8(4h,t,-ch2(ch3)-n-[ch2-ch2-ch2]2-),3.04(3h,s,-ch2(ch3)-n-[ch2-ch2-ch2]2-),3.18(8h,t,[cooch3-ch2-ch2]4-n),3.28(8h,t,[cooch3-ch2-ch2]4-n),3.62(12h,s,[cooch3-ch2-ch2]4-n)。
(2)化合物ii的制备
装置通氮气5min,然后取2.934g(6mmol)化合物ⅰ溶于4ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶内,再通氮气20min,取0.978(6mmol)6-溴-1己烯溶于3ml无水甲醇中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加结束后体系在25℃下反应96h。反应结束后,旋蒸除去甲醇,并用无水乙醚反复萃取提纯,最终得到3.14g黄色黏性液体即化合物ⅱ,收率为80.2%,由化合物ⅱ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)1.48(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.68(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.86(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.10(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.51(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.75(8h,t,[cooch3-ch2-ch2]4-n),3.0(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.25(3h,s,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.3(8h,t,[cooch3-ch2-ch2]4-n),3.66(12h,s,[cooch3-ch2-ch2]4-n),3.67(2h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),5.05(2h,t,ch2-ch=ch2),5.84(1h,m,ch2-ch=ch2)。
(3)化合物iii的制备
将装置置于冰水浴中,通氮气5min,然后取12.24g(120mmol)n,n-二甲基-1,3-丙二胺溶于15ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶中,在黑暗条件下搅拌并通氮气20min,取3.26g(5mmol)化合物ⅱ溶于5ml无水甲醇中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加完成后待体系自然恢复至室温,再设定温度25℃,黑暗条件下反应72h。旋蒸除去甲醇,并反复加入甲醇旋蒸除去过量的n,n二甲基1,3丙二胺,循环多次处理之后,最终得到3.85g淡黄色粘稠液体即化合物ⅲ,收率为82.7%,由化合物ⅲ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)1.41(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.62(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.82(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.15(24h,s,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.16(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.34(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.49(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.72(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.96(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),3.11(6h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.2(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),3.26(3h,s,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),5.0(2h,t,ch2-ch=ch2),5.8(1h,m,ch2-ch=ch2)。
(4)化合物iv的制备
装置通氮气5min,然后取4.482g(18mmol)1-溴十二烷溶于6ml无水丙酮中,加入上接球形冷凝管和干燥管的三口烧瓶中,再通氮气20min,取2.80g(3mmol)化合物ⅲ溶于9ml无水丙酮中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加完成设置35℃,反应80h。反应结束后,旋蒸除去丙酮,并用石油醚反复洗涤萃取,并用真空烘箱烘干,最终得到4.81g桔黄色颗粒状固体即化合物ⅳ,收率为83.2%,由化合物ⅳ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)0.82(12h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),1.21(88h,m,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),1.3(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.42(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.7(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),1.96(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.4(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.57(8h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),3.08(27h,s,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.25(14h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2,-ch2(ch3)n+-[ch2-ch2-ch2-n]2,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),3.35(8h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),5.0(2h,t,ch2-ch=ch2),5.8(1h,m,ch2-ch=ch2)。
实施例3
(1)化合物i的制备
搭建反应装置,包括水浴锅,三口烧瓶,球形冷凝管,干燥管,氮气供给装置。首先将搭建好的装置置于冰水浴中,并通氮气5min,取2.175g(15mmol)3,3-二氨基-n-甲基二丙胺并溶于15ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶内,再通氮气20min,然后量取9.03g(105mmol)丙烯酸甲酯在黑暗条件下以滴/3s的速度加入三口瓶中,滴加结束后待体系自然恢复至室温,然后设定40℃,反应36h。反应结束后,旋蒸除去甲醇,并反复加入无水甲醇旋蒸除去丙烯酸甲酯,循环5次以上,最终得到6.18g淡黄色微粘液体即化合物ⅰ,收率为84.3%,由化合物ⅰ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)2.15(4h,m,-ch2(ch3)-n-[ch2-ch2-ch2]2-),2.57(4h,t,-ch2(ch3)-n-[ch2-ch2-ch2]2-),2.8(4h,t,-ch2(ch3)-n-[ch2-ch2-ch2]2-),3.04(3h,s,-ch2(ch3)-n-[ch2-ch2-ch2]2-),3.18(8h,t,[cooch3-ch2-ch2]4-n),3.28(8h,t,[cooch3-ch2-ch2]4-n),3.62(12h,s,[cooch3-ch2-ch2]4-n)。
(2)化合物ii的制备
装置通氮气5min,然后取2.934g(6mmol)化合物ⅰ溶于4ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶内,再通氮气20min,取1.141(7mmol)6-溴-1己烯溶于3ml无水甲醇中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加结束后体系在40℃下反应87h。反应结束后,旋蒸除去甲醇,并用无水乙醚反复萃取提纯,最终得到3.26g黄色黏性液体即化合物ⅱ,收率为83.4%,由化合物ⅱ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)1.48(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.68(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.86(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.10(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.51(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.75(8h,t,[cooch3-ch2-ch2]4-n),3.0(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.25(3h,s,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.3(8h,t,[cooch3-ch2-ch2]4-n),3.66(12h,s,[cooch3-ch2-ch2]4-n),3.67(2h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),5.05(2h,t,ch2-ch=ch2),5.84(1h,m,ch2-ch=ch2)。
(3)化合物iii的制备
将装置置于冰水浴中,通氮气5min,然后取16.32g(160mmol)n,n-二甲基-1,3-丙二胺溶于15ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶中,在黑暗条件下搅拌并通氮气20min,取3.26g(5mmol)化合物ⅱ溶于15ml无水甲醇中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加完成后待体系自然恢复至室温,再设定温度26℃,黑暗条件下反应85h。旋蒸除去甲醇,并反复加入甲醇旋蒸除去过量的n,n二甲基1,3丙二胺,循环多次处理之后,最终得到3.97g淡黄色粘稠液体即化合物ⅲ,收率为85.3%,由化合物ⅲ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)1.41(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.62(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.82(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.15(24h,s,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.16(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.34(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.49(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.72(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.96(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),3.11(6h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.2(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),3.26(3h,s,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),5.0(2h,t,ch2-ch=ch2),5.8(1h,m,ch2-ch=ch2)。
(4)化合物iv的制备
装置通氮气5min,然后取5.229g(21mmol)1-溴十二烷溶于6ml无水丙酮中,加入上接球形冷凝管和干燥管的三口烧瓶中,再通氮气20min,取2.80g(3mmol)化合物ⅲ溶于9ml无水丙酮中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加完成设置40℃,反应96h。反应结束后,旋蒸除去丙酮,并用石油醚反复洗涤萃取,并用真空烘箱烘干,最终得到4.87g桔黄色颗粒状固体即化合物ⅳ,收率为84.2%,由化合物ⅳ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)0.82(12h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),1.21(88h,m,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),1.3(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.42(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.7(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),1.96(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.4(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.57(8h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),3.08(27h,s,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.25(14h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2,-ch2(ch3)n+-[ch2-ch2-ch2-n]2,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),3.35(8h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),5.0(2h,t,ch2-ch=ch2),5.8(1h,m,ch2-ch=ch2)。
实施例4
(1)化合物i的制备
搭建反应装置,包括水浴锅,三口烧瓶,球形冷凝管,干燥管,氮气供给装置。首先将搭建好的装置置于冰水浴中,并通氮气5min,取5.8g(40mmol)3,3-二氨基-n-甲基二丙胺并溶于15ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶内,再通氮气20min,然后量取17.2g(200mmol)丙烯酸甲酯在黑暗条件下以滴/3s的速度加入三口瓶中,滴加结束后待体系自然恢复至室温,然后设定35℃,反应28h。反应结束后,旋蒸除去甲醇,并反复加入无水甲醇旋蒸除去丙烯酸甲酯,循环5次以上,最终得到16.25g淡黄色微粘液体即化合物ⅰ,收率为83.1%,由化合物ⅰ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)2.15(4h,m,-ch2(ch3)-n-[ch2-ch2-ch2]2-),2.57(4h,t,-ch2(ch3)-n-[ch2-ch2-ch2]2-),2.8(4h,t,-ch2(ch3)-n-[ch2-ch2-ch2]2-),3.04(3h,s,-ch2(ch3)-n-[ch2-ch2-ch2]2-),3.18(8h,t,[cooch3-ch2-ch2]4-n),3.28(8h,t,[cooch3-ch2-ch2]4-n),3.62(12h,s,[cooch3-ch2-ch2]4-n)。
(2)化合物ii的制备
装置通氮气5min,然后取5.868g(12mmol)化合物ⅰ溶于14ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶内,再通氮气20min,取2.119(13mmol)6-溴-1己烯溶于3ml无水甲醇中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加结束后体系在35℃下反应77h。反应结束后,旋蒸除去甲醇,并用无水乙醚反复萃取提纯,最终得到6.50g黄色黏性液体即化合物ⅱ,收率为83.1%,由化合物ⅱ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)1.48(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.68(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.86(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.10(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.51(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.75(8h,t,[cooch3-ch2-ch2]4-n),3.0(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.25(3h,s,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.3(8h,t,[cooch3-ch2-ch2]4-n),3.66(12h,s,[cooch3-ch2-ch2]4-n),3.67(2h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),5.05(2h,t,ch2-ch=ch2),5.84(1h,m,ch2-ch=ch2)。
(3)化合物iii的制备
将装置置于冰水浴中,通氮气5min,然后取20.4g(200mmol)n,n-二甲基-1,3-丙二胺溶于15ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶中,在黑暗条件下搅拌并通氮气20min,取6.52g(10mmol)化合物ⅱ溶于25ml无水甲醇中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加完成后待体系自然恢复至室温,再设定温度28℃,黑暗条件下反应83h。旋蒸除去甲醇,并反复加入甲醇旋蒸除去过量的n,n二甲基1,3丙二胺,循环多次处理之后,最终得到8.02g淡黄色粘稠液体即化合物ⅲ,收率为86.0%,由化合物ⅲ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)1.41(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.62(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.82(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.15(24h,s,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.16(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.34(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.49(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.72(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.96(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),3.11(6h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.2(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),3.26(3h,s,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),5.0(2h,t,ch2-ch=ch2),5.8(1h,m,ch2-ch=ch2)。
(4)化合物iv的制备
装置通氮气5min,然后取13.446g(54mmol)1-溴十二烷溶于6ml无水丙酮中,加入上接球形冷凝管和干燥管的三口烧瓶中,再通氮气20min,取5.592g(6mmol)化合物ⅲ溶于9ml无水丙酮中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加完成设置38℃,反应78h。反应结束后,旋蒸除去丙酮,并用石油醚反复洗涤萃取,并用真空烘箱烘干,最终得到9.76g桔黄色颗粒状固体即化合物ⅳ,收率为84.4%,由化合物ⅳ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)0.82(12h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),1.21(88h,m,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),1.3(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.42(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.7(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),1.96(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.4(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.57(8h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),3.08(27h,s,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.25(14h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2,-ch2(ch3)n+-[ch2-ch2-ch2-n]2,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),3.35(8h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),5.0(2h,t,ch2-ch=ch2),5.8(1h,m,ch2-ch=ch2)。
实施例5
(1)化合物i的制备
搭建反应装置,包括水浴锅,三口烧瓶,球形冷凝管,干燥管,氮气供给装置。首先将搭建好的装置置于冰水浴中,并通氮气5min,取4.35g(30mmol)3,3-二氨基-n-甲基二丙胺并溶于10ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶内,再通氮气20min,然后量取18.06g(210mmol)丙烯酸甲酯在黑暗条件下以滴/3s的速度加入三口瓶中,滴加结束后待体系自然恢复至室温,然后设定37℃,反应34h。反应结束后,旋蒸除去甲醇,并反复加入无水甲醇旋蒸除去丙烯酸甲酯,循环5次以上,最终得到12.57g淡黄色微粘液体即化合物ⅰ,收率为85.7%,由化合物ⅰ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)2.15(4h,m,-ch2(ch3)-n-[ch2-ch2-ch2]2-),2.57(4h,t,-ch2(ch3)-n-[ch2-ch2-ch2]2-),2.8(4h,t,-ch2(ch3)-n-[ch2-ch2-ch2]2-),3.04(3h,s,-ch2(ch3)-n-[ch2-ch2-ch2]2-),3.18(8h,t,[cooch3-ch2-ch2]4-n),3.28(8h,t,[cooch3-ch2-ch2]4-n),3.62(12h,s,[cooch3-ch2-ch2]4-n)。
(2)化合物ii的制备
装置通氮气5min,然后取5.868g(12mmol)化合物ⅰ溶于14ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶内,再通氮气20min,取3.26(20mmol)6-溴-1己烯溶于3ml无水甲醇中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加结束后体系在32℃下反应80h。反应结束后,旋蒸除去甲醇,并用无水乙醚反复萃取提纯,最终得到6.67g黄色黏性液体即化合物ⅱ,收率为85.2%,由化合物ⅱ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)1.48(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.68(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.86(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.10(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.51(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.75(8h,t,[cooch3-ch2-ch2]4-n),3.0(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.25(3h,s,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.3(8h,t,[cooch3-ch2-ch2]4-n),3.66(12h,s,[cooch3-ch2-ch2]4-n),3.67(2h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),5.05(2h,t,ch2-ch=ch2),5.84(1h,m,ch2-ch=ch2)。
(3)化合物iii的制备
将装置置于冰水浴中,通氮气5min,然后取48.96g(480mmol)n,n-二甲基-1,3-丙二胺溶于25ml无水甲醇中,加入上接球形冷凝管和干燥管的三口烧瓶中,在黑暗条件下搅拌并通氮气20min,取6.52g(10mmol)化合物ⅱ溶于25ml无水甲醇中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加完成后待体系自然恢复至室温,再设定温度29℃,黑暗条件下反应90h。旋蒸除去甲醇,并反复加入甲醇旋蒸除去过量的n,n二甲基1,3丙二胺,循环多次处理之后,最终得到8.20g淡黄色粘稠液体即化合物ⅲ,收率为88.0%,由化合物ⅲ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)1.41(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.62(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.82(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.15(24h,s,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.16(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.34(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.49(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.72(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),2.96(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),3.11(6h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.2(8h,t,n-[ch2-ch2-conh-ch2-ch2-n(ch3)2]4),3.26(3h,s,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),5.0(2h,t,ch2-ch=ch2),5.8(1h,m,ch2-ch=ch2)。
(4)化合物iv的制备
装置通氮气5min,然后取13.446g(54mmol)1-溴十二烷溶于6ml无水丙酮中,加入上接球形冷凝管和干燥管的三口烧瓶中,再通氮气20min,取5.592g(6mmol)化合物ⅲ溶于9ml无水丙酮中,加入50ml恒压滴液漏斗中,以滴/3s的速度加入三口瓶中,滴加完成设置33℃,反应89h。反应结束后,旋蒸除去丙酮,并用石油醚反复洗涤萃取,并用真空烘箱烘干,最终得到9.81g桔黄色颗粒状固体即化合物ⅳ,收率为84.8%,由化合物ⅳ的核磁氢谱可知:1h-nmr(d2o,400mhz):δ(ppm)0.82(12h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),1.21(88h,m,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),1.3(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.42(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),1.7(4h,m,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),1.96(2h,m,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2),2.4(4h,t,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),2.57(8h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),3.08(27h,s,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4,-ch2(ch3)n+-[ch2-ch2-ch2-n]2),3.25(14h,t,-ch2(ch3)n+-ch2-ch2-ch2-ch2-ch=ch2,-ch2(ch3)n+-[ch2-ch2-ch2-n]2,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),3.35(8h,t,n-[ch2-ch2-conh-ch2-ch2-n+(ch3)2-(ch2)11-ch3]4),5.0(2h,t,ch2-ch=ch2),5.8(1h,m,ch2-ch=ch2)。
上述实施实例用来解释说明本发明,而不是对本发明进行限制,在本发明的精神和权力要求的保护范围内,对本发明做出的任何修改和改变,都落入本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!