一种以Fe-MOFs为前驱体可控制备单原子和原子簇铁催化剂的制作方法
本发明涉及无机功能材料制备技术领域,尤其涉及一种以fe-mofs为前驱体可控制备单原子和原子簇铁催化剂。
背景技术:
单原子催化(single-atomcatalysis,sac),可实现金属原子利用率的最大化,降低催化剂成本,因而具备极其重要的意义。相比于纳米/亚纳米催化剂,单原子催化剂具有诸多优势:(1)活性组分达到最大程度分散(100%),可有效提高金属原子利用率;(2)活性位点的组成和结构单一,可避免因活性组分组成和结构不均匀而导致的副反应,从而显著提高目标产物的选择性;(3)单原子催化剂兼具高活性、高选择性和可循环使用的优点,有望成为连接均相催化与非均相催化的桥梁。因此,单原子催化剂为在原子尺度上理解催化机理和构效关系提供了一个很好的平台。单原子催化剂同时具备均相催化剂“孤立位点”、多相催化剂结构稳定、易分离的优点,拥有桥连多相催化和均相催化的巨大潜力。
2011年,中科院大连化学物理研究所张涛院士领导的团队及合作者大胆突破传统思维,首次制备出单原子pt1/feox催化剂,并以co氧化为探针反应并结合理论计算详细研究了单原子金属催化的机理,并第一次在学术上提出“单原子催化”的观点,单原子金属催化受到科研工作者的热切关注。近年来,单原子金属催化更是魅力四射,大放异彩。例如,单原子金属催化剂可应用于co选择性氧化反应、甲烷无氧制乙烯及芳构化、水汽变换反应、芳香硝基化合物的选择性加氢反应。在单原子催化剂概念提出的短短几年里,它已经成为目前多相催化领域的研究热点,在催化领域展现出了非常巨大的潜力。我国多家单位课题组以及美国加州大学、加拿大西安大略大学、英国剑桥大学、阿斯顿大学、瑞士联邦理工学院、日本国家材料科学研究所等单位都相继开展了单原子金属催化剂的制备与催化性能研究工作,发展出许多新的单原子金属催化剂制备方法。但目前还没有一个比较普适的方法来合成单原子催化剂,也只有个别课题组通过不断的摸索在一些特殊的载体上能够合成出相对比较均匀的单分散的单原子催化剂。因此,单原子金属催化剂面临的首要挑战,是如何制备出孤立的并能稳定存在的单原子金属。由于单个原子具有较高的表面能,为避免团聚,制备的单原子金属催化剂负载量往往较低(<0.5wt%);另一方面,载体与金属单原子之间存在着微妙的金属-载体界面协同效应,但实际催化剂却存在着其界面精细结构表征难、构-效关系本质难被揭示的特点,这是单原子金属催化剂研究工作的另一个巨大挑战。
综上所述,现有技术中对于单原子金属催化剂的制备,尚缺乏有效的解决方案。
技术实现要素:
针对上述现有技术中存在的不足,本发明提供了一种单原子金属催化剂的可控制备方法。该方法利用fe-mofs为前驱体制备单原子和原子簇铁催化剂,降低了催化剂成本,提高了原子利用率,具有催化活性高,稳定性强等优点,以结构、组成可调控的负载型铁基单原子颗粒为模型催化剂来深入研究催化剂的复杂界面效应,这一策略也适用于其他催化体系的研究,将有助于开发更为高效、低廉的实用非贵金属催化剂。
本发明的目的是通过以下技术方案实现的:
a.按照铁盐与有机配体的摩尔比为0.5~4:1的比例,将两者混合后,再加入去离子水和十一烯酸,超声混合,制成混合反应液;
十一烯酸的添加,一方面可有效的提升fe-mofs前驱体的得率,另一方面,可在烧结前,起到保护fe-mofs前驱体的作用,防止其被环境中的氧化物质氧化,超声混合,则能有效的提升反应物之间的混合效果;
所述的铁盐是硝酸铁、硫酸铁、氯化铁、乙酰丙酮铁或乙酸铁中的任意一种;有机配体是富马酸;
所述去离子水与有机配体的体积摩尔比为0.4l:1mol,十一烯酸和有机配体的摩尔比为1:5~10,所述超声混合时,超声波的频率为32~38khz,超声处理的时间为10min。
b.将步骤a的混合反应液迅速转移到聚四氟乙烯反应釜中,以1~10℃/min的升温速率将反应物升温至80℃并恒温反应24h;反应结束后,自然冷却到室温,过滤出沉淀物、洗涤后置于40~80℃烘箱中干燥至恒重,得到含铁的金属有机骨架材料fe-mofs前驱体;
c.将步骤b得到的前驱体置于管式炉中,在惰性气体保护下以1~10℃/min的升温速率升温至300~600℃并恒温处理3h,再以5℃/min的升温速率升温至900℃恒温2h,冷却至室温取出即可;所述的惰性气体是氮气或氩气中的任意一种。
本发明的有益效果为:
本发明以含铁的金属有机骨架材料为前驱体,经高温热解法制备得到了一种负载在碳上的单原子和原子簇铁催化材料(fe-sa-c),该方法降低了催化剂制备成本,提高了原子利用率,具有催化活性高,稳定性强等优点。制备得到的单原子和原子簇铁催化剂中金属原子的负载量为5%~20%,非贵金属原子利用效率能达到99.5%以上。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域的普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他附图。
图1为本发明所制得的fe-mofs和fe-sa-c材料的扫描电镜照片。
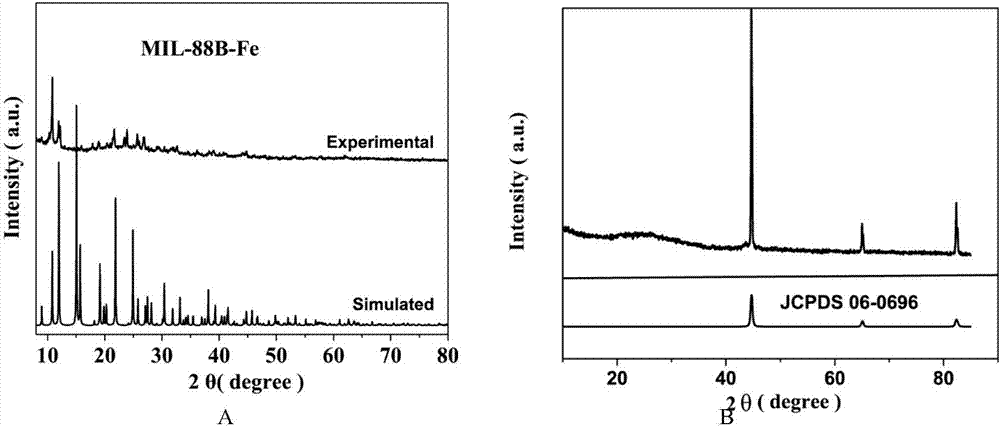
图2为本发明所制得的fe-mofs和fe-sa-c材料的x-射线粉末衍射对比图谱。
图3为本发明所制得的fe-sa-c材料的透射电镜照片。
具体实施方式
下面结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明的实施例,本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明的保护范围。
下面对本发明所提供的一种以fe-mofs为前驱体可控制备单原子和原子簇铁催化剂进行详细描述。
(一)一种fe-mofs前驱体
一种fe-mofs前驱体,如图1(a)所示其微观形貌为纺锥体,直径在500nm~1μm,长度在5μm。
(二)上述fe-mofs前驱体的制备方法
一种fe-mofs前驱体的制备方法,包括以下步骤:
步骤a、按照铁盐与有机配体的摩尔比为0.5~4:1的比例,将两者混合后,再加入去离子水和富马酸,超声混合,制成混合反应液,所述去离子水与有机配体的体积摩尔比为0.4l:1mol,十一烯酸和有机配体的摩尔比为1:5~10,所述超声混合时,超声波的频率为32~38khz,超声处理的时间为10min;
步骤b、将步骤a的混合反应液迅速转移到聚四氟乙烯反应釜中,以1~10℃/min的升温速率将反应物升温至60~100℃并恒温反应6~24h;
步骤c、反应结束后,自然冷却到室温,离心、洗涤后置于40~80℃烘箱中干燥至恒重,得到含铁的金属有机骨架材料(fe-mofs)前驱体;
(三)一种fe-sa-c材料
一种fe-sa-c材料,如图1(b)所示的微观形貌是纺锥体,直径在500nm~1μm,长度在5~10μm。
(四)上述fe-sa-c材料的制备方法
一种fe-sa-c材料的制备方法,包括以下步骤:
将上述(二)中的步骤c得到的前驱体置于管式炉中,在惰性气体保护下以1~10℃/min的升温速率升温至300~600℃并恒温处理3h,再以5℃/min的升温速率升温至900℃恒温2h,冷却至室温取出即可。
由上述本发明提供的技术方案可以看出,本发明所提供的纺锥体fe-mofs前驱体的制备方法先采用六水合三氯化铁、富马酸和去离子水在水热条件下直接合成了纺锥体fe-mofs前驱体,然后将该纺锥体fe-mofs前驱体在惰性气体下分段煅烧得到了fe-sa-c材料。由此可见,本发明所提供的fe-sa-c材料可能具有良好的催化性能,而且成本低廉、原料丰富、制备方法简单,适合大规模生产应用。
为了更加清晰地展现出本发明所提供的技术方案及所产生的技术效果,下面以具体实施例对本发明实施例所提供的以fe-mofs为前驱体可控制备单原子和原子簇铁催化剂进行详细描述。
实施例1
一种fe-mofs前驱体的制备方法,包括以下步骤:
步骤a、按照每2.703g的六水合三氯化铁使用1.161g的富马酸、400ml的去离子水、0.369g十一烯酸,将六水合三氯化铁、富马酸、去离子水、十一烯酸超声混合10min,制成混合反应液;
步骤b、将步骤a的混合反应液迅速转移到聚四氟乙烯反应釜中,以5℃/min的升温速率将反应物升温至80℃并恒温反应12h;
步骤c、反应结束后,自然冷却到室温,离心、洗涤后置于60℃烘箱中干燥至恒重,得到含铁的金属有机骨架材料(fe-mofs)前驱体。
实施例2
一种fe-sa-c材料的制备方法,包括以下步骤:
将上述(二)中的步骤c得到的前驱体置于管式炉中,在惰性气体保护下以5℃/min的升温速率升温至400℃并恒温处理3h,再以5℃/min的升温速率升温至900℃恒温2h,冷却至室温取出即可。
所述的惰性气体是氮气。
由上述本发明提供的技术方案可以看出,本发明所提供的纺锥体fe-mofs前驱体的制备方法先采用六水合三氯化铁、富马酸和去离子水在水热条件下直接合成了纺锥体fe-mofs前驱体,然后将该纺锥体fe-mofs前驱体在惰性气体下分段煅烧得到了fe-sa-c材料。由此可见,本发明所提供的fe-sa-c材料可能具有良好的催化性能,而且成本低廉、原料丰富、制备方法简单,适合大规模生产应用。
实施例1和实施例2的表征检测
(1)采用扫描隧道显微镜分别对本发明实施例1和实施例2所制得的fe-mofs和fe-sa-c材料进行检测,从而得到如图1所示的纺锥体,大小均一。
(2)采用x-射线粉末衍射仪分别对本发明实施例1和实施例2所制得的fe-mofs和fe-sa-c材料进行检测,从而得到如图2所示的x-射线粉末衍射对比图谱(即xrd对比图谱)。
(3)采用场发射透射电子显微镜(tecnaig2f20)对实施例2所制得的fe-sa-c材料的微观形貌,从而得到如图3所示的,图3(a)是低倍的透射照片中可以看出fe-sa-c材料依然保持着纺锥体,图3(b)中可以看出选区内没有铁的颗粒,图3(c)是其透射扫描照片,从图中可以清晰的看出存在着单分散的铁原子和铁原子簇。
综上可见,本发明实施例成功制备出fe-sa-c材料可能具有良好的催化性能,而且成本低廉、原料丰富、制备方法简单,适合大规模生产应用。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求书的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!