毕赤酵母基因敲除和抗性基因回收载体及构建方法与运用与流程
本发明属于基因工程领域,更具体地,涉及一种毕赤酵母基因敲除的方法。
背景技术:
毕赤酵母(pichiapastoris)是一类能以甲醇作为唯一碳源和能量来源进行生长的甲醇营养型酵母。毕赤酵母含有醇氧化酶aox1强启动子,这是目前调控机理最严格的启动子之一,在甲醇为唯一碳源时,基因的表达可受甲醇严格调控。毕赤酵母表达系统因其生长速度快、易于基因遗传转化操作、有多种强启动子可选择、表达效率高、可实现高密度发酵等优点而被广泛应用于各种外源蛋白质的表达,并获得了较高的蛋白表达水平。
毕赤酵母体内无天然质粒存在,一般将载体整合入基因组内以形成稳定遗传的转化子。为了实现外源蛋白的高效表达或揭示某些未知基因的功能,往往需要对宿主进行一系列的遗传操作以实现相应的生物学目的。其中,基因敲除作为一种常用的技术手段,被广泛的应用于毕赤酵母的研究。
普遍使用的基因敲除手段是通过同源重组双交换方式实现基因的插入失活或基因删除。在传统的酿酒酵母中进行基因敲除,往往只需要50bp左右的同源臂就能实现有效的基因敲除。然而,在一些非传统酵母如毕赤酵母中,由于非同源末端连接修复机制导致基因敲除过程中双交换事件发生的概率降低,为了实现相对高效的敲除,一般使用长度大于1000bp的同源臂,对于某些基因的敲除甚至需要超过2000bp的同源臂,尽管能够实现敲除的目的,但敲除的成功率仍然相对较低。
目前,实现无抗性基因残留的遗传操作主要采取的是抗生素基因回收策略。主要包括基于位点特异性重组法(如cre-loxp、flp-frt等重组系统)和利用自杀基因作为反筛选标记剔除法(如ura3基因在ura3营养缺陷型酵母作为自杀基因、大肠杆菌毒素蛋白mazf等)。然而利用ura3反向筛选标记,需要使用尿嘧啶缺陷型菌株,极大地限制了其应用范围。
目前,主要有以下几种方法应用于提高毕赤酵母基因敲除的效率。(1)通过增加同源臂的长度,提高同源重组双交换发生的概率;(2)敲除毕赤酵母中ku70基因,通过破坏其非同源重组末端连接途径,使得酵母内的基因修复主要通过同源重组途径,从而提高敲除效率。然而非同源重组作为毕赤酵母体内重要的基因修复途径之一,其功能的缺陷可能会造成宿主的稳定性降低或其他不可预知的缺陷;(3)通过构建辅助载体预先表达待敲除基因,通过增加基因的冗余弥补基因敲除后可能导致的生长缺陷实现基因的高效敲除。
尽管结合上述各种方法能实现毕赤酵母基因的敲除,然而毕赤酵母基因敲除的成功率仍然偏低,某些难敲除基因(比如α-1,6-甘露糖转移酶基因och1等)的敲除往往需要筛选大量的克隆子而导致敲除过程十分困难,甚至得不到敲除成功的克隆子;或者在实现提高敲除效率的同时,残留了标记基因,无法实现标记基因的重复利用,同时,多种筛选标记的残存往往会留下潜在的遗传风险。
因此,构建一种高效毕赤酵母基因敲除方法的同时实现敲除后标记基因的回收,具有重要的应用价值。
技术实现要素:
针对现有技术中毕赤酵母基因敲除的成功率低、抗性敲除效率与基因敲除后筛选标记回收效率难以兼顾、使用的同源臂较长、敲除过程目的克隆子筛选工作量大等技术缺陷或改进需求,本发明提供了一种毕赤酵母基因敲除和抗性基因回收载体及构建方法与运用。
为实现上述目的,按照本发明的一个方面,提供了一种毕赤酵母基因敲除的模板载体paoxz-mazf,该模板载体包括毕赤酵母甲醇型启动子aox1调控的mazf基因表达盒和博来霉素抗性基因表达盒;
所述mazf表达盒的5’端含有用于插入待敲除基因下游同源片段的酶切位点a,该酶切位点的5’端含有cre特异性重组系统识别的lox66位点;
所述mazf表达盒的3’端含有用于插入待敲除基因上游同源片段的酶切位点b,该酶切位点的3’端含有cre特异性重组系统识别的lox71位点;
所述博来霉素抗性基因表达盒位于所述lox66位点的5’端与所述lox71位点的3’端之间;
所述酶切位点a和酶切位点b为不同的酶切位点,且酶切位点a和酶切位点b在所述模板载体上唯一存在。
按照本发明的另一方面,提供了一种毕赤酵母基因敲除的模板载体paoxz-mazf的构建方法,包括以下步骤:
(1)在大肠杆菌中扩增得到mazf基因,在mazf基因的两端引入酶切位点;
(2)将步骤(1)得到的引入了酶切位点的mazf基因酶切后,得到暴露出粘性末端的mazf基因;将载体ppicza经过酶切后,得到暴露出与mazf基因相同粘性末端的ppicza载体;将所述暴露出粘性末端的mazf基因与暴露出相同粘性末端的ppicza载体进行混合连接,将该连接了mazf基因的ppicza载体转化大肠杆菌感受态细胞,经含有博来霉素的平板筛选后,随机挑选长出的克隆子抽质粒酶切验证后,经测序验证后命名为ppicza-mazf;
(3)以步骤(2)得到的ppicza-mazf为模板,使用引物lox66-f和lox71-r1第一轮pcr扩增得到含有lox66位点和mazf表达盒的片段,将该片段作为第二轮pcr的模板,使用引物lox66-f和lox71-r2通过第二轮pcr扩增得到两端分别含有lox66和lox71位点的mazf表达盒片段;
(4)将步骤(3)得到的片段酶切后纯化回收,然后与ppicza-mazf载体酶切后纯化回收含有抗生素基因部分的片段连接后,转化大肠杆菌感受态细胞,经含有博来霉素的平板筛选,随机挑选长出的克隆子抽质粒酶切验证后,经测序验证后命名为paoxz-mazf。
优选地,步骤(1)所述的酶切位点是ecori和sali酶切位点;步骤(4)所述将步骤(3)得到的片段酶切是采用bamhi单酶切;步骤(4)所述ppicza-mazf载体酶切是bglii和bamhi双酶切。
按照本发明的另一方面,提供了所述毕赤酵母基因敲除的模板载体用于敲除毕赤酵母基因的应用。
优选地,所述应用包括以下步骤:
(1)目的基因敲除载体的构建:根据待敲除的基因序列的下游序列设计整合同源臂,根据待敲除的基因序列的上游序列设计敲除同源臂;所述整合同源臂作为同源片段将敲除载体整合入毕赤酵母基因组;所述敲除同源臂用于基因敲除过程中mazf基因表达盒丢失的同源片段;将所述整合同源臂和敲除同源臂分别插入权利要求1所述的paoxz-mazf载体的酶切位点a和b处,其中,整合同源臂的5’端为lox66位点,敲除同源臂的3’端为lox71位点,构成目的基因敲除载体;所述整合同源臂含有至少一个所述目的基因敲除载体上唯一存在的酶切位点;
(2)目的基因敲除载体整合入毕赤酵母基因组:将步骤(1)所述目的基因敲除载体在整合同源臂片段处线性化回收,转入毕赤酵母菌株感受态细胞,在含有博来霉素抗性平板涂布筛选,得到的克隆子进一步经过基因组pcr验证确认所述目的基因敲除质粒整合入目的基因下游位点处;
(3)将步骤(2)中确认正确的克隆子接种于含有博来霉素抗性的甲醇诱导液体培养基中培养到菌体浑浊,再将菌体划线接种于毕赤酵母生长培养基平板分离得到单菌落,对分离得到的单菌落进行pcr验证,得到已完成目的基因敲除的毕赤酵母;
优选地,步骤(3)所述的毕赤酵母生长培养基平板为含有博来霉素抗性的平板。
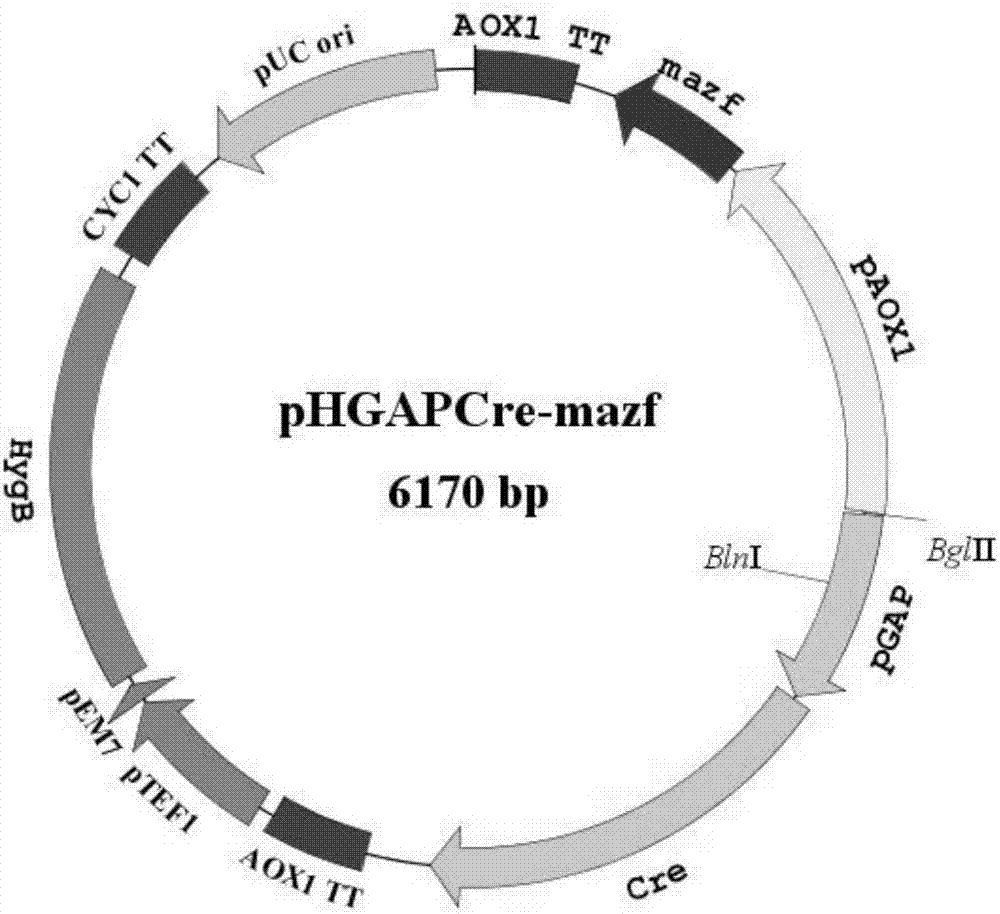
按照本发明的另一方面,提供了一种毕赤酵母基因抗性基因回收载体phgapcre-mazf,该回收载体包括:利用毕赤酵母甲醇型启动子aox1调控的mazf基因表达盒aox1-mazf-aox1tt、利用gap启动子组成型表达的cre基因表达盒gap-cre-aox1tt和hygb抗性基因表达盒tef1-hygb-cyctt;所述mazf基因表达盒和cre基因表达盒在所述phgapcre-mazf回收载体上方向相反;所述gap启动子处含有至少一个在所述phgapcre-mazf回收载体唯一存在的酶切位点。
按照本发明的另一方面,提供了一种毕赤酵母基因抗性基因回收载体phgapcre-mazf的构建方法,包括以下步骤:
(1)将hygb片段、tef1启动子片段和cyctt片段经过重叠延伸pcr融合连接得到tef1-hygb-cyctt表达盒,将该tef1-hygb-cyctt表达盒和pgapzαa载体酶切纯化后连接得到以潮霉素b作为筛选标记的毕赤酵母表达载体pgaphαa;
(2)将cre基因片段与步骤(1)得到的pgaphαa载体经过酶切后回收纯化得到的载体片段通过重组连接得到表达载体phgap-cre;
(3)将权利要求2所述的ppicz-mazf载体通过酶切纯化后得到含有aox1-mazf-aox1tt表达盒后插入步骤(2)得到的phgap-cre载体后,筛选aox1-mazf-aox1tt表达盒与gap-cre-aox1tt表达盒反向连接的载体得到phgapcre-mazf。
优选地,步骤(1)所述的酶切是bamhi和mlui双酶切;步骤(2)所述的酶切是asuii和noti双酶切;步骤(2)所述的酶切是bamhi和bglii双酶切。
按照本发明的另一方面,提供了所述毕赤酵母基因抗性基因回收载体phgapcre-mazf在回收毕赤酵母基因抗性基因方面的应用。
优选地,所述应用,包括以下步骤:
(1)将权利要求书6所述的毕赤酵母基因抗性基因回收载体phgapcre-mazf整合入毕赤酵母基因组;将phgapcre-mazf在gap启动子位点处线性化回收后,转入权利要求5中所述已完成目的基因敲除的毕赤酵母感受态细胞,在含有潮霉素b抗生素的平板涂布筛选,得到的克隆子进一步经过基因组pcr验证,确认phgapcre-mazf在gap启动子位点处插入已完成目的基因敲除的毕赤酵母基因组;
(2)博来霉素抗性基因的回收:将步骤(1)得到的转入phgapcre-mazf载体的毕赤酵母分别接种于含有和不含有博来霉素的抗性平板进行抗性平板验证,对不具有博来霉素抗性的克隆子进一步通过基因组pcr验证确认组成型表达的cre重组酶已实现lox71和lox66位点之间的重组剪接;
(3)抗性基因回收载体phgapcre-mazf的回收:将步骤(2)确认组成型表达的cre重组酶已实现lox71和lox66位点之间的重组剪接的克隆子接种于甲醇诱导液体培养基中培养到菌体浑浊,再将菌体划线接种于毕赤酵母生长培养基平板,对分离得到的单菌落分别接种于含有和不含有潮霉素b的抗性平板进行验证hygb抗性,对不具有hygb抗性的克隆子进一步通过基因组pcr验证所述phgapcre-mazf载体实现完全回收,即得到无抗性敲除的毕赤酵母菌株。
相对于现有技术,本发明具有以下有益效果:
(1)本发明极大地提高了毕赤酵母基因的敲除效率。本发明通过构建一种基于mazf自杀基因的敲除载体,线性化后电转化入待敲除毕赤酵母感受态细胞,得到的重组菌几乎都是以单交换方式整合入指定待敲除基因下游同源片段位点的克隆子,极大的避免了传统毕赤酵母敲除过程中由于同源重组双交换或非同源末端修复等因素导致的特定整合方式效率低下。在敲除过程中,通过诱导mazf自杀基因表达同时加以抗生素筛选这种双向筛选,引导mazf表达盒在指定的同源片段处发生交换而自我剔除同时导致目的基因的敲除,阳性率极高,大大提高了筛选得到基因敲除克隆的效率。
(2)本发明提供了基因敲除后抗生素标记回收的方法,为多基因的高效敲除提供了极大的便利。为了实现敲除后抗性基因回收的目的,以便于多基因的敲除。通过转入phgapcre-mazf质粒,载体整合到酵母基因组gap启动子位点的同时,通过利用gap组成型启动子控制的cre重组酶基因,实现诱导敲除过程的引入的筛选标记的回收。敲除过程引入的抗生素标记完成回收后,通过诱导mazf基因自杀,实现phgapcre-mazf载体的去除,最终实现基因的无抗性敲除。且抗生素基因的回收的步骤都可以通过抗生素耐受性对菌体快速鉴别得到目的克隆子,筛选过程十分简单高效。
附图说明
图1为敲除模板载体paoxz-mazf的结构示意图;
图2为抗性回收载体phgapcre-mazf的结构示意图;
图3为paoxz-mazf敲除载体实现基因敲除过程原理图;
图4为敲除载体结构示意图。其中图a为gas1基因敲除载体paoxz-mazf(gas1)的结构示意图,图b为och1基因敲除载体paoxz-mazf(och1)的结构示意图;
图5为敲除gas1基因过程中菌株基因组pcr验证电泳检测图。其中图a为gs115/paoxz-mazf(gas1)菌株进行ppic-f和gout-r引物对pcr验证;图b为gs115/△gas1-zeocin菌株进行gas1-f和gas1-r引物对验证,;图c为gs115/△gas1-zeocin菌株回收抗性前后基因组验证;
图6为gs115/△gas1-paoxz-mazf(och1)克隆子在含有100μg/mlzeocin的mmh液体培养基中摇瓶生长3-4天后划线接种于ypd平板的生长状况;
图7为敲除och1基因过程中菌株基因组pcr验证电泳检测图。其中图a为gs115/△gas1-paoxz-mazf(och1)克隆子进行ppic-f和oout-r引物对pcr验证;图b为gs115/△gas1-△och1-zeocin克隆子pcr验证结果;
图8为phgapcre-mazf回收载体实现抗性基因回收过程原理图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
实施例中所用的菌株和质粒:大肠杆菌dh5α感受态细胞自制并保存;pgapzαa、ppicza、gs115购于invitrogen公司。
酶和试剂盒等:限制性内切酶、pcr所用的酶制剂、连接酶试剂盒solutioni等购于takara公司;treliefsosoocloningkit重组连接试剂盒购于擎科生物有限公司;质粒提取试剂盒和dna回收试剂盒购于omega公司。
生长条件:大肠杆菌于37℃生长,毕赤酵母菌于28℃生长。
合成引物:
实施例1:一种毕赤酵母基因敲除的模板载体paoxz-mazf,如图1所示,该模板载体包括毕赤酵母甲醇型启动子aox1调控的mazf基因表达盒和博来霉素抗性基因表达盒;
所述mazf表达盒的5’端含有用于插入待敲除基因下游同源片段的酶切位点a,该酶切位点的5’端含有cre特异性重组系统识别的lox66位点;
所述mazf表达盒的3’端含有用于插入待敲除基因上游同源片段的酶切位点b,该酶切位点的3’端含有cre特异性重组系统识别的lox71位点;
所述博来霉素抗性基因表达盒位于所述lox66位点的5’端与所述lox71位点的3’端之间;所述酶切位点a和酶切位点b为不同的酶切位点,且酶切位点a和酶切位点b在所述模板载体上唯一存在。
实施例2:一种毕赤酵母基因抗性基因回收载体phgapcre-mazf,如图2所示,该回收载体包括:利用毕赤酵母甲醇型启动子aox1调控的mazf基因表达盒aox1-mazf-aox1tt、利用gap启动子组成型表达的cre基因表达盒gap-cre-aox1tt和hygb抗性基因表达盒tef1-hygb-cyctt;所述mazf基因表达盒和cre基因表达盒在所述phgapcre-mazf回收载体上方向相反;所述gap启动子处含有至少一个在所述phgapcre-mazf回收载体唯一存在的酶切位点。
实施例3:毕赤酵母基因敲除的模板载体paoxz-mazf的构建
(1)以大肠杆菌dh5α菌体为模板,使用引物mazf-f和mazf-r通过菌落pcr扩增得到mazf基因,在基因的两端引入ecori和sali酶切位点,琼脂糖凝胶电泳后回收纯化;
(2)以ecori和sali双酶切后纯化回收,然后与商业化载体ppicza经过ecori和sali酶切(去除了载体自身的xhoi酶切位点)后胶回收的载体以3:1混合,通过takara公司的连接试剂盒solutioni在16℃下连接1h后转化大肠杆菌dh5α感受态细胞,克隆子经含有25μg/mlzeocin的llb平板筛选,随机挑选长出的克隆子抽质粒酶切验证后,经测序验证正确后命名为ppicza-mazf;
(3)以ppicza-mazf为模板,使用引物lox66-f和lox71-r1第一轮pcr扩增得到含有lox66位点和mazf表达盒的片段,琼脂糖凝胶电泳回收后作为第二轮pcr的模板,使用引物lox66-f和lox71-r2通过第二轮pcr扩增得到两端分别含有lox66和lox71位点的mazf表达盒片段;
(4)上一步得到的片段经过bamhi单酶切后纯化回收,然后与ppicza-mazf载体经过bglii和bamhi双酶切后纯化回收含有抗生素基因部分的片段连接,转化大肠杆菌dh5α感受态细胞,克隆子经含有25μg/mlzeocin的llb平板筛选,随机挑选长出的克隆子抽质粒酶切验证后,经测序筛选验证,mazf表达盒的方向保持与ppicza-mazf一致的克隆子命名为paoxz-mazf。
实施例4:毕赤酵母基因抗性基因回收载体phgapcre-mazf的构建
(1)以商业化载体pgapzαa为模板,分别用tef-f/tef-r和cyc-f/cyc-r引物对pcr得到tef启动子和cyc终止子部分片段;
(2)以本实验室保存的pcmbia1301载体为模板,以hyg-f和hyg-r引物对pcr扩增hyg基因部分;
(3)tef启动子片段和hyg基因片段1:1混合后作为模板,以tef-f和hyg-r引物重叠延伸pcr扩增得到tef启动子和hyg基因融合片段;
(4)tef启动子和hyg基因融合片段和cyc终止子片段1:1混合后作为模板,以tef-f和cyc-r引物重叠延伸pcr扩增得到hygb基因表达盒片段后纯化回收;
(5)hygb基因表达盒片段与pgapzαa载体通过bamhi和mlui双酶切纯化后以3:1混合,通过takara公司的连接试剂盒solutioni在16℃下连接1h后转化大肠杆菌dh5α感受态细胞,克隆子经含有100μg/ml潮霉素b的lb平板筛选,随机挑选长出的克隆子抽质粒酶切验证后,经测序筛选验证正确的命名为pgaphαa;
(6)以本实验室保存的cre+t载体为模板,以cre-f和cre-r引物对pcr后回收纯化得到的cre基因片段与pgaphαa载体经过asuii和noti双酶切后回收纯化得到的载体片段以2:1混合后,通过重组连接酶2×sosoo在50℃下重组连接15min后转化大肠杆菌dh5α感受态细胞,克隆子经含有100μg/ml潮霉素b的lb平板筛选,克隆子经过cre-f和cre-r引物对菌落pcr筛选后,经测序筛选验证正确的命名为pgaph-cre;
(7)ppicz-mazf载体通过bamhi和bglii双酶切回收纯化得到含有mazf表达盒片段与phgap-cre载体通过bglii单酶切回收的载体片段以3:1混合后,通过takara公司的连接试剂盒solutioni在16℃下连接1h后转化大肠杆菌dh5α感受态细胞,克隆子经含有100μg/ml潮霉素b的lb平板筛选,随机挑选长出的克隆子抽质粒酶切验证后,经测序筛选“aox1-mazf-aox1tt”表达盒与“gap-cre-aox1tt”表达盒反向的载体验证正确的命名为phgapcre-mazf。
实施例5:毕赤酵母gas1基因的无抗性敲除
毕赤酵母基因敲除的模板载体paoxz-mazf。如图3所示,paoxz-mazf质粒以博来霉素抗性筛选;paoxz-mazf用于载体整合入指定待敲除基因位点后,通过甲醇诱导mazf表达,大肠杆菌的mazf基因的编码产物通过特异识别和切割mrna的5’-aca-3’序列,对宿主产生致死效果,同时施加zeocin抗性选择压力,宿主为了存活迫使mazf基因表达盒以同源重组方式丢失的同时实现基因的高效敲除,基因敲除后基因组处残留zeocin筛选标记,且筛选标记两侧分别含有cre特异性重组酶特异性识别位点lox71和lox66,根据需要可用于筛选标记的回收重复利用。
毕赤酵母基因抗性基因回收载体phgapcre-mazf。如图8所示,phgapcre-mazf质粒以潮霉素b抗性筛选;含有用gap启动子调控组成型表达的cre位点特异性重组酶,用于组成型诱导第一轮基因敲除后在基因组残留的lox71和lox66位点之前重组剪接,实现第一轮敲除引入的zeocin筛选标记的回收,同时在基因组遗留重组后形成的lox72痕迹,且不再被cre重组酶识别;含有aox1诱导的mazf表达盒,在甲醇诱导下,mazf的毒性迫使phgapcre-mazf在以gap启动子的两端同源片段间发生敲除事件,用于完成第一轮抗性标记回收后phgapcre-mazf载体的自我剔除。
本实施例采用毕赤酵母基因敲除的模板载体paoxz-mazf和毕赤酵母基因抗性基因回收载体phgapcre-mazf用于敲除毕赤酵母gas1基因的无抗性敲除,实施过程包括以下步骤:
(1)以毕赤酵母gs115基因组为模板,以upgas1-f和upgas1-r引物对pcr后回收纯化得到的gas1基因上游同源片段即敲除同源臂与paoxz-mazf载体经过xhoi单酶切后回收纯化得到的载体片段以2:1混合后,通过重组连接酶2×sosoo在50℃下重组连接15min后转化大肠杆菌dh5α感受态细胞,克隆子经含有25μg/mlzeocin的llb平板筛选,克隆子经过upgas1-f和upgas1-r引物对菌落pcr筛选后,经测序筛选验证正确的命名为paoxz-mazf(upgas1);
(2)以毕赤酵母gs115基因组为模板,以dwgas1-f和dwgas1-r引物对pcr后回收纯化得到的gas1基因下游同源片段即整合同源臂与paoxz-mazf(upgas1)载体经过bglii单酶切后回收纯化得到的载体片段以2:1混合后,通过重组连接酶2×sosoo在50℃下重组连接15min后转化大肠杆菌dh5α感受态细胞,克隆子经含有25μg/mlzeocin的llb平板筛选,克隆子经过dwgas1-f和dwgas1-r引物对菌落pcr筛选后,经测序筛选验证正确的命名为paoxz-mazf(gas1)(如附图4a所示);
(3)paoxz-mazf(gas1)载体经过xbai单酶切线性化后电转化入毕赤酵母gs115感受态细胞,重组子经含有100μg/mlzeocin的ypds平板筛选。生长出的克隆子随机挑选3个抽取基因组,通过ppic-f和gout-r引物对pcr验证,结果表明3个克隆子都是符合预期整合方式的阳性克隆子,即paoxz-mazf(gas1)载体在gs115基因组gas1基因下游同源片段位点处插入的重组子,命名为gs115/paoxz-mazf(gas1);
(4)gs115/paoxz-mazf(gas1)克隆子接种于含有100μg/mlzeocin的mmh液体培养基中摇瓶生长3-4天后划线接种于ypd平板。随机挑选2个在ypd平板生长出的单克隆抽取基因组,通过gas1-f和gas1-r引物对以及gout-f和tef-r引物对pcr验证,验证结果表明挑选的2个毕赤酵母重组子都实现了gas1基因的敲除,重组子命名为gs115/△gas1-zeocin。
(5)抗性回收载体phgapcre-mazf经过avrii线性化纯化回收后,电转化入gs115/△gas1-zeocin感受态细胞,重组子经含有300μg/ml潮霉素b的ypds平板筛选。在潮霉素b抗生素平板生长出的克隆子分别点板至ypd平板和含有100μg/mlzeocin的ypd平板,实验表明约80%重组子丢失zeocin抗性。丢失zeocin抗性的克隆子抽取基因组经gout-f和gout-r引物对pcr验证后进一步经过测序分析表明,gas1基因实现敲除而且zeocin抗性实现了回收,phgapcre-mazf回收载体中所述mazf基因表达盒和cre基因表达盒在所述phgapcre-mazf回收载体上方向相反为了避免两个终止子同向,避免在两个终止子之间发生敲除事件,而不能实现抗性基因的敲除。同时gas1基因位点处仅残留lox71和lox66位点重组后生成的lox72疤痕,重组子命名为gs115/△gas1-hygb,基因型验证结果如附图5所示,其中图a为gs115/paoxz-mazf(gas1)菌株进行ppic-f和gout-r引物对pcr验证,其中泳道1为gs115基因组,作为阴性对照,泳道2、3分别为gs115/paoxz-mazf(gas1)1#、2#克隆子基因组,泳道4为水,作为空白对照;图b为gs115/△gas1-zeocin菌株进行gas1-f和gas1-r引物对验证,其中泳道1为gs115/paoxz-mazf(gas1)1#基因组,作为阴性对照,泳道2、3分别为gs115/△gas1-zeocin1#、2#克隆子基因组,泳道4为水,作为空白对照;图c为gs115/△gas1-zeocin菌株回收抗性前后基因组验证,其中1-4泳道为gs115/△gas1-zeocin菌株进行gout-f和tef-r引物对验证,泳道1为gs115基因组,作为阴性对照,泳道2、3分别为gs115/△gas1-zeocin1#、2#克隆子基因组,泳道4为水,作为空白对照,泳道5-8为gs115/△gas1菌株进行gout-f和gout-r引物对验证,泳道5为gs115基因组,作为阴性对照,泳道6、7分别为gs115/△gas11#、2#菌株基因组,泳道8为水,作为空白对照。从图5中可以看出,其中图a证明了paoxz-mazf(gas1)载体正确插入gs115基因组gas1基因下游同源片段位点处;图b结果表明gs115/△gas1-zeocin1#、2#菌株不再含有gas1基因,即已实现gas1基因敲除;图c结果表明gs115/△gas11#、2#在成功实现gas1基因敲除的基础上成功实现zeocin标记的回收。
(6)gs115/△gas1-hygb克隆子接种于含有mmh液体培养基中摇瓶生长3-4天后划线接种于ypd平板。ypd平板长出的单克隆分别点板至ypd平板和含有300μg/ml潮霉素b的ypd平板,几乎所有重组子丢失潮霉素b抗性,表明gs115/△gas1-hygb重组子实现了phgapcre-mazf载体的回收,所得到的重组子即成功实现了gas1基因的无抗性敲除,命名为gs115/△gas1。
实施例6:毕赤酵母菌gs115/△gas1进一步进行och1基因敲除
(1)以毕赤酵母gs115基因组为模板,以upoch1-f和upoch1-r引物对pcr后回收纯化得到的och1基因上游同源片段与paoxz-mazf载体经过xhoi单酶切后回收纯化得到的载体片段以2:1混合后,通过重组连接酶2×sosoo在50℃下重组连接15min后转化大肠杆菌dh5α感受态细胞,克隆子经含有25μg/mlzeocin的llb平板筛选,克隆子经过upoch1-f和upoch1-r引物对菌落pcr筛选后,经测序筛选验证正确的命名为paoxz-mazf(upoch1);
(2)以毕赤酵母gs115基因组为模板,以dwoch1-f和dwoch1-r引物对pcr后回收纯化得到的och1基因下游同源片段与paoxz-mazf(upoch1)载体经过bglii单酶切后回收纯化得到的载体片段以2:1混合后,通过重组连接酶2×sosoo在50℃下重组连接15min后转化大肠杆菌dh5α感受态细胞,克隆子经含有25μg/mlzeocin的llb平板筛选,克隆子经过dwoch1-f和dwoch1-r引物对菌落pcr筛选后,经测序筛选验证正确的命名为paoxz-mazf(och1)(如附图4b所示);
(3)paoxz-mazf(och1)载体经过psti单酶切线性化后电转化入毕赤酵母gs115/△gas1感受态细胞,重组子经含有100μg/mlzeocin的ypds平板筛选。生长出的克隆子随机挑选4个抽取基因组,通过ppic-f和oout-r引物对pcr验证,结果表明4个克隆子都是符合预期整合的阳性克隆子,即paoxz-mazf(och1)载体在gs115/△gas1基因组och1基因下游同源片段位点处插入的重组子,命名为gs115/△gas1-paoxz-mazf(och1);
(4)gs115/△gas1-paoxz-mazf(och1)克隆子接种于含有100μg/mlzeocin的mmh液体培养基中摇瓶生长3-4天后划线接种于ypd平板。约有超过70%克隆子生长十分缓慢且长出的酵母菌表面褶皱,如附图6所示,平板划线分离单克隆的过程中,最初划的几条线毕赤酵母菌浓度较高,很多菌株都无法分离,此外生长状态正常的菌株相比敲除och1基因的菌株生长速度快且菌落很大,所以很多密集菌落里面即使混入了很多成功敲除och1基因的菌株也无法判断,故计数范围选取菌株分离开的那部分单菌落。这些符合och1基因敲除后毕赤酵母生长特性的克隆子,暗示其och1基因实现敲除。随机挑选4个在ypd平板生长出的疑似och1基因被敲除的克隆子抽取基因组,通过och1-f和och1-r引物对、oout-f和oout-r引物对以及oout-f和cyc-r引物对pcr验证,验证结果表明挑选的4个毕赤酵母重组子都实现了och1基因的敲除,重组子命名为gs115/△gas1-△och1-zeocin(基因型验证结果如附图7所示)。其中图a为gs115/△gas1-paoxz-mazf(och1)克隆子进行ppic-f和oout-r引物对pcr验证,其中泳道1为gs115基因组,作为阴性对照,泳道2-5分别为gs115/△gas1-paoxz-mazf(och1)1#-4#克隆子基因组;图b为gs115/△gas1-△och1-zeocin克隆子pcr验证结果,其中泳道1-5为och1-f和och1-r引物对pcr验证,泳道1为gs115/△gas1-paoxz-mazf(och1)1#,作为阴性对照,泳道2-5分别为gs115/△gas1-△och1-zeocin1#-4#菌株基因组,泳道6-10为oout-f和oout-r引物对pcr验证,泳道6为gs115/△gas1-paoxz-mazf(och1)1#,作为阴性对照,泳道7-10分别为gs115/△gas1-△och1-zeocin1#-4#菌株基因组,泳道11-15为oout-f和cyc-r引物对pcr验证,泳道11为gs115基因组,作为阴性对照,泳道12-15分别为gs115/△gas1-△och1-zeocin1#-4#菌株基因组。从图7可以看出,其中图a结果表明paoxz-mazf(och1)载体正确插入gs115/△gas1基因组och1基因下游同源片段位点处;图b结果综合表明gs115/△gas1-△och1-zeocin1#-4#菌株不再含有och1基因,即实现了och1基因的敲除。
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
<110>华中科技大学
<120>毕赤酵母基因敲除和抗性基因回收载体及构建方法与运用
<160>30
<170>siposequencelisting1.0
<210>1
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>1
cggaattcacgatggtaagccgatacgtac30
<210>2
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>2
acgcgtcgacctacccaatcagtacgttaa30
<210>3
<211>60
<212>dna
<213>人工序列(artificialsequence)
<400>3
gggatccataacttcgtatagcatacattatacgaacggtagatctaacatccaaagacg60
<210>4
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>4
cggtactcgaggcacaaacgaaggtctcac30
<210>5
<211>57
<212>dna
<213>人工序列(artificialsequence)
<400>5
cgggatccataacttcgtataatgtatgctatacgaacggtactcgaggcacaaacg57
<210>6
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>6
cgggatcccccacacaccatagc23
<210>7
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>7
ggtgagttcaggctttttcatggtttagttcctcaccttgtc42
<210>8
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>8
gacaaggtgaggaactaaaccatgaaaaagcctgaactcacc42
<210>9
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>9
cgtgggccgccgtcggacgtgctatttctttgccctcggacg42
<210>10
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>10
cgtccgagggcaaagaaatagcacgtccgacggcggcccacg42
<210>11
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>11
cgacgcgtctgtacagaaaaaaaag25
<210>12
<211>52
<212>dna
<213>人工序列(artificialsequence)
<400>12
caattgaacaactatttcgaaacggaattcatgtccaatttactgaccgtac52
<210>13
<211>49
<212>dna
<213>人工序列(artificialsequence)
<400>13
gagtttttgttctagaaagctggcggccgcttacaaacagcttccttcg49
<210>14
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>14
ggtagctcttgatccggcaaac22
<210>15
<211>26
<212>dna
<213>人工序列(artificialsequence)
<400>15
cggaattcatgtttaaatctctgtgc26
<210>16
<211>32
<212>dna
<213>人工序列(artificialsequence)
<400>16
atttgcggccgcttagaatgacataatcattc32
<210>17
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>17
gagaccttcgtttgtgcctcgagcgatctctggctgatccgag43
<210>18
<211>49
<212>dna
<213>人工序列(artificialsequence)
<400>18
gtatgctatacgaacggtactcgagattttgattatctttgtgagacgc49
<210>19
<211>47
<212>dna
<213>人工序列(artificialsequence)
<400>19
gcatacattatacgaacggtagatctgcggttcacattaattaaagg47
<210>20
<211>45
<212>dna
<213>人工序列(artificialsequence)
<400>20
cctttcgtctttggatgttagatctgaagcacaagacaggcccag45
<210>21
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>21
ccacatggggttcagtcctac21
<210>22
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>22
ggatccattattggagaagaag22
<210>23
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>23
atggcgaaggcagatggcag20
<210>24
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>24
ttagtccttccaacttccttc21
<210>25
<211>44
<212>dna
<213>人工序列(artificialsequence)
<400>25
gagaccttcgtttgtgcctcgagtggtagggatgcaatacaagg44
<210>26
<211>44
<212>dna
<213>人工序列(artificialsequence)
<400>26
gtatgctatacgaacggtactcgagggctgatgatatttgctac44
<210>27
<211>46
<212>dna
<213>人工序列(artificialsequence)
<400>27
gcatacattatacgaacggtagatctagaaagctagagtaaaatag46
<210>28
<211>45
<212>dna
<213>人工序列(artificialsequence)
<400>28
cctttcgtctttggatgttagatctcgaggttggttggtcaatgg45
<210>29
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>29
gtcacacacggagaagtaatg21
<210>30
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>30
cacgaattccttgtggaagatc22
- 还没有人留言评论。精彩留言会获得点赞!