氨解方法及氨解组合物与流程
本发明涉及生物工程领域,尤其是与dna固相合成相关的氨解方法。本发明还涉及用于氨解的氨解组合物。
背景技术:
dna合成(尤其是dna引物合成)是大多是基于固相合成技术,通过脱保护、缩合、氧化以及盖帽四个步骤的循环,将亚磷酰胺单体按3’端到5’端的顺序逐个连接至控制孔径玻璃球(cpg)表面。每个亚磷酰胺单体的5’端羟基由二甲氧基三苯基(dmt)基团保护,其碱基上的活性氨基分别由乙酰基(ac)、苯甲酰基(bz)、二氮甲基甲脒基(dmf)等基团保护。其中dmt基团为酸不稳定的,可在3%三氯乙酸的二氯甲烷等溶液中被切除,ac、bz、dmf等基团为碱不稳定基团,可在饱和氨水等溶液中被切除。
合成的具体步骤为:
a)使用三氯乙酸溶液将cpg表面的dmt基团切除,露出自由羟基;
b)以四氮唑为活化剂,使亚磷酰胺单体活化的3’端磷酸基团与cpg表面的亚磷酰胺单体的5’端羟基缩合,形成磷酸二酯键;
c)使用碘溶液作为氧化剂,将引物骨架中不稳定的三价磷氧化成稳定的五价磷;
d)使用乙酸酐与氮甲基咪唑作为盖帽试剂,将步骤b)中未参与反应的自由羟基乙酰化;
e)重复以上a)至d)步骤直至序列合成结束,并脱去最后一个dmt基团。
待合成结束后,将cpg粉末浸泡在饱和氨水溶液中并放置在55℃进行氨解反应。该过程主要包含三步反应:i)将引物从cpg上切除;ii)将各碱基上的氨基保护基团切除;iii)将3’端的磷酸基团切除。在合成时,若3’端需进行磷酸化修饰,可以通过选择不同类型的cpg而对iii)步骤进行选择性省略。
使用饱和氨水进行氨解时,通常需要在55℃反应16小时,或在65℃反应8小时。使用饱和氨水与甲胺水溶液的1:1混合液(ama)作为氨解试剂,可以将反应时间可以缩短至15分钟左右。但由于上述氨解方式需将cpg粉末浸没在氨解溶液中而容易引起交叉污染,且在氨解之后需要花2小时左右将溶液抽干,故在引物的大规模工业生产应用中存在一定困难。
现有的微波氨解方式可将氨解反应时间降低至1小时左右。该微波氨解方式需将cpg粉末浸泡于氢氧化钠的水和甲醇混合液中,在微波条件下进行反应。由于温度升高可能导致溶液爆沸,故需要每隔6-10秒便将反应管取出置于冰水中冷却1-2秒,待反应结束后再将溶液抽干得到最终的产物。该方法存在以下缺点:i)氨解试剂在过程中容易挥发而造成可重复性较差;ii)反复冷却,操作过于繁琐,整体耗时较长;iii)反应结束后引物无法进行清洗,且抽干水和甲醇混合液耗时较长;iv)当使用96孔板或384孔板进行大规模引物合成时,兼容性较差,容易产生交叉污染。
即使二代引物合成柱使用cpg与基质制成的熔块(即cpgfrits)代替cpg粉末作为固相载体,通过提升合成过程中的试剂渗透性而提升了引物合成质量,但该类合成柱中的固相载体在氨解时较难取出而浸没于氨解试剂中,也降低了微波氨解的工业化可行性。
使用气相氨解的方式可显著降低交叉污染的可能。该氨解方式需将引物合成柱放置在高压容器中,使用气体氨气为氨解试剂,在高压(约140psi)下反应30分钟左右。虽然该方法后处理环节较为简单,但是该氨解方式对于设备的需求较高。
技术实现要素:
为了解决上述问题,在一方面,本发明提供了一种用于在dna固相合成后进行氨解的方法,其包括以下步骤:
1)向包括用于合成dna片段的固相载体的合成柱内加入氨解组合物;
2)将所述合成柱置于装有氨解缓冲液的可密封容器内,并保持所述合成柱位于所述氨解缓冲液的液面上方;以及
3)以微波处理所述可密封容器,使得所述固相载体处于所述氨解缓冲液的蒸汽气氛中;
4)取出所述合成柱,以洗脱液处理所述固相载体并收集流出液。
在一个实施方案中,所述固相载体为cpg粉末或cpgfrits。
在一个实施方案中,所述合成柱的数量为一个。在另一个实施方案中,所述合成柱的数量为一个以上。
在一个实施方案中,所述可密封容器内可设置有可用于夹持所述合成柱的支持物。
在另一实施方案中,所述合成柱位于合成柱支架上。相应地,可在所述可密封容器内设置用于托住所述合成柱支架的支持物。在优选的实施方案中,所述合成柱支架为96孔板支架或384孔板支架。
在一个实施方案中,以体积计,步骤1)中所述氨解组合物的用量为所述固相载体体积的0.3至5倍。
在一个实施方案中,步骤1)还包括通过离心或加压让加入的所述氨解组合物在所述合成柱内流动,从而遍布于所述合成柱的固相载体内。
在一个优选的实施方案中,步骤1)中在上述离心或加压处理后还包括向所述合成柱内补加所述氨解组合物并静置1分钟以上。优选地,补加的氨解组合物的量为固相载体体积的0.3至5倍。
在一个实施方案中,步骤2)中还包括让所述合成柱在所述可密封容器内保持倒置状态。
在一个具体实施方案中,步骤3)中所述微波处理包括以700w微波处理5至20分钟。
在一个实施方案中,步骤4)中在所述洗脱液处理之前还包括通过离心和/或烘干和/或微波加热除去所述合成柱上残留的小水滴。在优选的实施方案中,在离心除去所述小水滴时让所述合成柱以倒置状态离心。
在一个实施方案中,步骤4)中在所述洗脱液处理之前还包括以清洗试剂清洗所述合成柱一次或更多次。优选地,所述清洗试剂为甲醇水溶液或乙腈。优选地,所述甲醇水溶液为85%(v/v)甲醇水溶液。
在一个实施方案中,所述洗脱液为水,优选无菌水。
在一个实施方案中,所述氨解组合物为1:2-20:5-40体积比的二亚乙基三胺、1m碱性水溶液和醇的混合溶液。优选地,所述碱性水溶液选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化铍、氢氧化镁、氢氧化钙溶液及其组合。在一个实施方案中,所述氨解缓冲液为1:5-80体积比的醇和水的混合溶液。优选地,所述醇选自甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇、仲丁醇、叔丁醇及其组合。
在一个实施方案中,所述dna片段长度为10-200个碱基。
在一个实施方案中,所述dna片段长度为10-100个碱基。
在一个实施方案中,所述dna片段长度为10-60个碱基。
在一个实施方案中,所述dna片段为引物。
另一方面,本发明还提供了氨解组合物,其包括1:2-20:5-40体积比的二亚乙基三胺、1m碱性水溶液和醇。优选地,所述碱性水溶液选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化铍、氢氧化镁、氢氧化钙溶液及其组合。优选地,所述醇选自甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇、仲丁醇、叔丁醇及其组合。
另一方面,本发明还提供了所述氨解组合物在氨解反应中的用途,包括让所述氨解组合物与完成了dna片段合成的固相载体接触。
与现有的氨解方式相比,本发明提供的氨解方法仅需使用较少量的氨解试剂,可避免交差污染,缩短了反应时间,方便清洗溶剂清洗,并且无需高压设备,有利于工业化和自动化使用。
附图说明
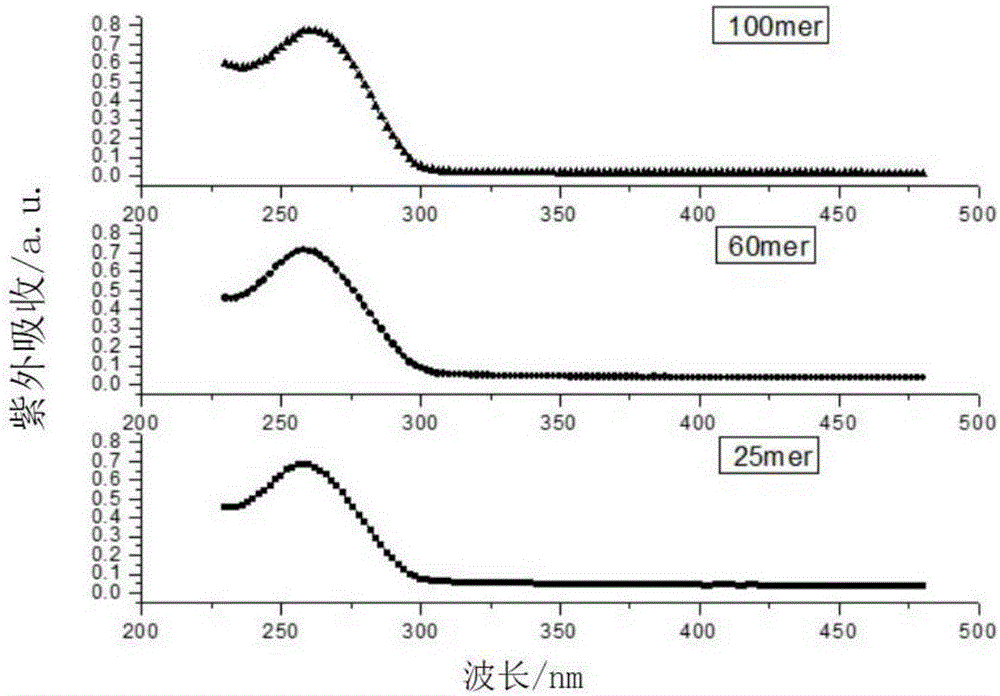
图1显示了经本发明氨解方法处理的不同长度引物的紫外光谱。
图2显示了经本发明氨解方法处理的不同长度引物的高效液相色谱图。
图3显示了经本发明氨解方法处理的25个碱基引物的质谱图。
图4显示了经本发明氨解方法处理的60个碱基引物的质谱图。
图5显示了经本发明氨解方法处理的100个碱基引物的质谱图。
图6显示了经传统氨解方法处理的不同长度引物的紫外光谱。
图7显示了经传统氨解方法处理的不同长度引物的高效液相色谱图。
具体实施方式
除非另有说明,本发明所用的技术和科学术语具有本发明所属领域的普通技术人员通常所理解的含义。
本文所用的术语“dna固相合成”指本领域内常用的一种合成dna链的方法,包括:将所要合成的dna链的末端核苷酸(例如,3’末端核苷酸)预先固定在一种不溶性固相载体上,再从此末端开始将其它核苷酸按预定顺序逐一接长,直至合成整个dna链。每接长一个核苷酸残基经历一轮相同的操作(见背景技术部分),并且保持延伸中的dna链始终固定在固相载体上。过量的未反应物或反应副产物可通过过滤或洗涤的方法除去。合成至所需长度后的dna链可从固相载体上切割下来并脱去各种保护基,再经纯化即可得到最终产物。以这种方式合成的dna链通常为几十个碱基,或者多至几百个碱基,它们可以例如用作pcr引物、接头或探针等。
本文所用的术语“合成柱”通常包括固相载体、筛板和空柱管三部分。空柱管通常为聚丙烯注塑而成,上口比下口大,固相载体和筛板装填于其中。常用的固相载体为粉末状的控制孔径玻璃球(controlledporeglass,cpg)。cpg球体内部有很多不规则的孔道,流动相可在其中流动。筛板通常用uhmw-pe或hdpe粉末烧结而成。uhmw-pe粉末在一定的温度下处于半融化状态,颗粒之间搭桥形成一定的孔径。在dna合成中,筛板的作用是阻挡cpg不漏下来而合成试剂能够通过。目前更常使用的是“第二代合成柱”,此种合成柱由cpgfrits(含控制孔径玻璃球的滤芯)和空柱管构成。cpgfrits是由cpg和uhmw-pe或hdpe粉末烧结成。uhmw-pe颗粒包裹并固定cpg颗粒,uhmw-pe颗粒之间搭桥形成一定的孔径。流动相可在cpg内部的孔和uhmw-pe颗粒之间的孔中流动,完成各种化学反应(参见公开号为cn204151332u的中国专利)。
本文在提及合成柱时使用的术语“倒置”或“倒置状态”指,相对于合成柱上口在上、下口在下的常见状态,将合成柱以上口在下、下口在上的方式放置在支架或者支持物上。
本文使用的术语“氨解(ammonolysis)”或“氨解反应”指,在dna固相合成的最后阶段,通过氨水溶液(或者其它碱性溶液)将合成的dna链从固相载体上切下、将碱基上的氨基保护基团切除和/或将3’末端的磷酸基团切除的化学过程。
本文使用的术语“氨解组合物”指用于进行上述氨解过程的组合物,也可以称为氨解试剂。在本发明的一些实施方案中,该氨解组合物为1:2-20:5-40体积比的二亚乙基三胺、1m碱性水溶液和醇的混合溶液。该碱性水溶液例如为氢氧化锂、氢氧化钠、氢氧化钾、氢氧化铍、氢氧化镁、氢氧化钙溶液等。所用的醇一般为低级醇,例如,甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇、仲丁醇、叔丁醇等。
本文使用的术语“可密封容器”指用于在其中进行氨解反应的容器,该容器可以例如通过加盖而进行密封。该可密封容器是微波可穿透的,例如由玻璃、陶瓷、塑料等制成。在进行氨解反应时,预先加入其中氨解缓冲液在微波处理下产生蒸汽。蒸汽通过合成柱的上口和下口而进入固相载体的孔径内(即让合成柱处于“蒸汽气氛”中),促进氨解组合物与合成的dna链进行的氨解反应。
本文所用的术语“微波”指频率为300mhz至300ghz、即波长在1毫米至1米之间的电磁波。对于玻璃、塑料和瓷器,微波几乎是穿透而不被吸收的。水和含水溶液会吸收微波而使自身发热。金属类物质则会反射微波。在本发明的一些实施方案中,可以采用微波炉来产生微波。通常,微波炉产生的微波的频率在2450mhz左右。
本发明提供的氨解方法,包括以下步骤:
1)向包括用于合成dna片段的固相载体的合成柱内加入氨解组合物;
2)将合成柱置于装有氨解缓冲液的可密封容器内,并保持合成柱位于氨解缓冲液的液面上方;
3)以微波处理所述可密封容器,使得固相载体处于氨解缓冲液的蒸汽气氛中;以及
4)取出合成柱,以洗脱液处理固相载体并收集流出液。
在步骤1)的一些实施方案中,向合成柱内加入少量的氨解组合物,例如为固相载体体积的0.3至5倍,例如例如0.5、0.6、0.8、1、2、3倍。为了让氨解组合物能够有效地分布于固相载体内而充分参与氨解反应,可以通过离心或者加压的方式,让作为流动相的氨解组合物沿固相载体流动,从而遍布于固相载体。在一些优选的实施方案中,为了防止在上述离心或加压处理过程中过多的氨解组合物移动至固相载体的下部(甚至从固相载体的下部流出),导致固相载体的上部氨解组合物的量过少,可以向合成柱内补加适量的氨解组合物,并静置一段时间,例如2-10分钟。补加的氨解组合物的量可以与之前添加的量相同或不同。
在步骤2)的一些实施方案中,可密封容器内可放置有适合于夹持合成柱的支持物。这种情况下,合成柱直接放置在支持物上,并且支持物上有适合于夹持合成柱的孔或者其它结构。在另一些情形下,可以将合成柱放置在合成柱支架(例如,96孔板或384孔板支架)上,再将该合成柱支架放置在可密封容器内的支持物上。这些支持物可以保持合成柱位于氨解缓冲液的液面上方,以免合成柱浸泡在氨解缓冲液中。对支持物的形状、大小或者结构无特别要求,只要适合于夹持合成柱或者托住合成柱支架即可。在本发明的优选实施方案中,在进行氨解反应时让合成柱处于倒置状态(即让合成柱倒置在支持物上),以方便蒸汽从下往上进入固相载体内。
在步骤3)的一些实施方案中,通过微波炉产生的微波处理可密封容器。所用的微波功率可以为200w至1000w,例如500w、600w、700w、800w等。该微波处理一方面可以在可密封容器中产生蒸汽,让合成柱处于蒸汽气氛中,另一方面可促进氨解组合物与合成的dna链之间的氨解反应。在本发明的一些实施方案中,微波处理的时间为1至20分钟,例如5分钟、6分钟、7分钟、8分钟、9分钟、10分钟、11分钟、12分钟、13分钟、14分钟、15分钟等。显然,微波处理时间与所用的微波功率、可密封容器以及期望的氨解反应完成程度有关,本领域技术人员可容易地通过试验而获得适合的微波功率和处理时间组合。
在步骤4)的一些实施方案中,取出合成柱后,先以倒置状态离心去除合成柱上残留的小水滴(由蒸汽凝结产生),再进行后续的洗脱步骤。在另一些实施方案中,可以通过烘干或者在微波炉中以微波加热来去除残留的小水滴。在一些实施方案中,在洗脱之前可以用清洗试剂清洗合成柱,以去除未反应的试剂和反应副产物。常用的清洗试剂包括甲醇水溶液(优选85%(v/v)甲醇水溶液)或乙腈等。可以用这些清洗试剂清洗一次或多次。
本发明人发现,1:2-20:5-40体积比的二亚乙基三胺、1m碱性水溶液和醇的氨解组合物具有较好的氨解效果。超出此配比范围的其它组成也可能具有某种程度的氨解效果,但同时可能会有其它不利影响,例如,碱溶液配制的浓度太高时会产生溶解性的问题,即会产生沉淀,并且碱溶液比例较高时,后来洗脱出的dna片段(引物)在质谱检测时容易产生盐峰。又例如,可能会造成氨解产率偏低(od值较小)、引物最后经抽干后外观发白(由盐导致)、以及氨解效率偏低(各保护基团脱保护不完全等)等情况。
下面将结合具体实施例对本发明的一些方面进行详细描述。除非另有说明,下文描述的实施例的方法和材料均为可以通过购买获得的常规产品。
实施例1
本实施例的目的是合成长度为25个、60个以及100个碱基的引物,序列分别为:
5’-aaacccagggcctcaaggacaaacc-3’(目标分子量为7623.0)
5’-
ttctttcctccctaaacactgacctcccgcagtcttcactagcaggtgaggtcacctgta-3’(目标分子量为18232.8)
5’-
gtaccgagctcggatccgccaccatggctgaaaatagtgtattaacatccactactgggaggactagcttggcagactcttccatttttgattctaaagt-3’(目标分子量为30782.0)
1.引物合成
使用50nmol合成柱(购自北京迪纳兴科生物科技有限公司,c1001-50),四氮唑为活化试剂,3%(v/v)三氯乙酸/二氯甲烷为脱保护试剂,20%(v/v)乙酸酐/乙腈、16%(v/v)氮甲基咪唑/乙腈为盖帽试剂,0.025m碘液为氧化试剂。在dr.oligo192合成仪上合成相应的引物序列。
2.微波氨解
1)将引物合成柱转移至96孔板支架上,并向合成柱内加入20μl微波氨解试剂(二亚乙基三胺:1m氢氧化锂水溶液:乙醇=1:3:6,体积比);
2)将96孔板支架正面向上,在300rpm/min转速离下离心1分钟(离心机购自上海卢湘仪离心机仪器有限公司,型号l-550),并补加20μl微波氨解试剂,随后静置3分钟;。
3)向微波氨解玻璃槽(尺寸17cm×12cm×6cm)中加入80ml微波缓冲液,并将96孔板支架倒置于氨解支架上,盖紧氨解槽盒盖,放入700w微波炉中高火反应12分钟;
4)反应结束后,将96孔板支架倒置于离心机中,300rpm/min离心1分钟;
5)将96孔板支架侧放在微波炉中加热1分钟,随后在通风橱内冷却5分钟;
6)向各合成柱中加入200μl乙腈,1600rpm/min离心5-10秒,弃去流出液,并重复一次;
7)向各合成柱中加入200μl85%甲醇的水溶液,1600rpm/min离心1分钟,弃去流出液;
8)向各合成柱中加入200μl乙腈,1600rpm/min离心1分钟,弃去流出液;
9)将96孔板支架放置在96孔深孔板上,向各合成柱内加入200μl灭菌水,静置1分钟后,1600rpm/min离心1分钟,收集流出液。
10)将收集的引物溶液进行hplc、ms以及uv分析,结果分别显示在图1至图5中。由图1可见,三种不同长度的引物均呈现较好的紫外光谱,在260nm处有明显的紫外吸收。由图2可见,三种不同长度的引物在hplc上呈现较好的纯度。由图3至图5可见,三种不同长度的引物均在质谱上显示出正确的分子量。以上数据说明本发明提供的微波氨解组合物能较好地应用于各种长度引物的氨解。
实施例2
本实施例的目的是合成长度为25个、60个以及100个碱基的引物,使用传统的微波氨解,并将结果与本发明提供的微波氨解的组合物进行对比。合成的序列分别为:
5’-aaacccagggcctcaaggacaaacc-3’(目标分子量为7623.0)
5’-
ttctttcctccctaaacactgacctcccgcagtcttcactagcaggtgaggtcacctgta-3’(目标分子量为18232.8)
5’-
gtaccgagctcggatccgccaccatggctgaaaatagtgtattaacatccactactgggaggactagcttggcagactcttccatttttgattctaaagt-3’(目标分子量为30782.0)
1.引物合成
使用50nmol的合成柱,四氮唑为活化试剂,3%(v/v)三氯乙酸/二氯甲烷为脱保护试剂,20%(v/v)乙酸酐/乙腈、16%(v/v)氮甲基咪唑/乙腈为盖帽试剂,0.025m碘液为氧化试剂。在dr.oligo192合成仪上合成相应的引物序列。
2.传统微波氨解
1)将合成柱内cpg与基质的熔块取出并放入15ml离心管中,加入4ml氨解液(0.2mnaoh,水:甲醇=1:4,v/v),旋紧管盖并密封;
2)使用微波输出功率为700w微波炉微波加热6s,立即取出放于冰水中冷却1s,重复上述操作共40个循环;
3)反应结束后,向氨解液中加入50μl醋酸中和反应,使用speedvac抽干;
4)将引物溶于200μl灭菌水,并进行hplc、ms以及uv分析,结果显示在图6和图7中。
由图6可见,三种长度的引物经传统微波氨解后均在260nm处呈明显的紫外特征吸收峰,但其230nm处亦有明显杂质吸收。由图7可见,三种长度的引物经传统微波氨解后在hplc上呈现的纯度较差,存在明显分裂峰以及杂峰。此外,三种长度的引物经传统微波氨解后均无法通过质谱得到正确的分子量。由此可见,经由传统微波氨解方法得到的引物质量较差,且操作繁杂,较难用于工业化生产。
在本发明方法的实施方案中,氨解组合物(或氨解试剂)的用量通常为合成柱内固相载体体积的0.3至5倍。在现有技术中,如果采用较少量的氨解组合物,则氨解组合物会在微波处理过程中过快地挥发,难以有效地完成氨解反应。本发明人通过在氨解反应的密封环境中提供氨解缓冲液的蒸汽而巧妙地解决了这个问题。
本发明方法的一些明显优势包括但不限于,减少了氨解组合物用量,降低了成本;氨解过程无爆沸问题,无需反复冷却,操作简单;无需将多个合成柱浸没在氨解溶液中,避免了交叉污染;无需抽干氨解溶液,缩短了氨解时间。在本发明方法的实施方案中,也不需要将cpg粉末或者cpgfrits从合成柱内取出,所以方便后续的清洁溶剂洗涤。另外,与采用高压设备的气相氨解相比,本发明方法所需的装置(可密封容器、微波炉等)显然更为简单。由此,显然地,本发明提供的氨解方法可以用于大规模地工业化和自动化生产。
本发明所属领域技术员应理解,以上描述的方法和材料,仅是示例性的,而不应视为限定本发明的范围。
- 还没有人留言评论。精彩留言会获得点赞!