基因SOX10作为靶点在制备延缓黑色素瘤适应性耐药的药物中的应用的制作方法
本发明属于生物医药技术领域,涉及基因SOX10作为靶点在制备延缓黑色素瘤适应性耐药的药物中的应用。
背景技术:
近年来,随着环境污染、臭氧层破坏等原因,黑色素瘤日益成为严重威胁人类健康和生命的重大疾病之一,其治疗药物和手段的研发一直是生命科学领域研究的重点。
BRAF基因是RAF家族中的一员,编码一种丝/苏氨酸蛋白激酶。据研究表明,40-60%的欧美黑色素瘤患者携带BRAF激酶突变,其中第600号密码子缬氨酸到谷氨酸的突变(BRAFV600E)占大约90%。BRAFV600E突变造成 BRAF激酶异常活化并持续激活RAF/MEK/ERK信号通路,导致细胞增殖失调,细胞对凋亡的抗性增加,从而促进肿瘤的发生。BRAF基因的高突变率以及与多种肿瘤发生的密切关系使之成为小分子靶向药物的一个理想靶点。其中RAF抑制剂Vemurafenib已通过美国FDA认证,用于治疗携带BRAFV600E突变的晚期黑色素瘤病人,且取得了良好的治疗效果。从临床上看,RAF抑制剂在短期内对BRAF突变的肿瘤有着良好的抑制效果,但绝大多数的病人在长期治疗后都产生了耐药性。肿瘤细胞对小分子药物的初始敏感度受到肿瘤的适应性耐药能力的影响。
与经过药物长期选择产生的获得性耐药不同,适应性耐药是指肿瘤细胞对小分子药物在初始阶段就发生的应激反应,通过迅速、可逆的重构与生存相关的信号通路使肿瘤细胞在小分子药物作用下存活更长的时间,直至永久性的获得性耐药发生。最新研究发现BRAF突变肿瘤细胞在被RAF抑制剂处理后能应激性地迅速提高FOXD3的表达水平进而激活其下游的 ERBB3/PI3K/AKT信号通路,从而发生对RAF抑制剂的适应性耐受。然而,FOXD3是如何被RAF抑制剂快速诱导表达的这一关键科学问题尚未得到解答。
技术实现要素:
本发明解决的问题在于提供基因SOX10作为靶点在制备延缓黑色素瘤适应性耐药的药物中的应用。
本发明是通过以下技术方案来实现:
基因SOX10作为靶点在制备延缓黑色素瘤适应性耐药的药物中的应用。
所述的药物是促使SOX10表达下调以抑制FOXD3的表达。
所述的药物是使SOX10基因的T240和/或T244位点的磷酸化的药物,该药物能抑制FOXD3的转录活性。
所述的药物是促使SOX10表达下调以促进RAF抑制剂抑制BRAF突变黑色素瘤。
所述的药物是促使SOX10表达下调以抑制FOXD3/ERBB3/AKT信号通路并促进RAF抑制剂诱导的细胞凋亡。
所述的药物是调节FOXD3表达的药物,SOX10通过调控NRG1/ ERBB3/AKT信号通路的活性来调节FOXD3的表达。
所述的药物是延缓ERBB3介导的适应性耐药。
与现有技术相比,本发明具有以下有益的技术效果:
本发明通过实验验证了在BRAF突变肿瘤病人组织中SOX10和FOXD3 表达水平呈正相关,并揭示了SOX10是RAF抑制剂诱导FOXD3表达的充分必要条件;而且RAF抑制剂介导的适应性耐药中ERK1/2可以通过磷酸化 SOX10 T240/T244位点抑制其对FOXD3及其它下游靶基因的转录活性,充分表明SOX10基因是抑制剂如何诱导FOXD3表达、进而产生适应性耐药的关键调控因子。而且进一步实验表明SOX10表达下调可以有效抑制 FOXD3/ERBB3/AKT信号通路并显著促进抑制剂诱导的细胞凋亡、以及SOX10表达下调显著促进RAF抑制剂抑制肿瘤的效果并显著抑制肿瘤组织中FOXD3/ERBB3/AKT信号通路,因此基因SOX10可以作为靶点在制备延缓黑色素瘤适应性耐药的药物中进行应用,以制备更好更多的延缓黑色素瘤适应性耐药的药物。
附图说明
图1为SOX10是RAF抑制剂诱导FOXD3表达的充分必要条件。a为黑色素瘤细胞转染非靶向siRNA(对照)和SOX10特异性siRNA72小时,2 μM Vemurafenib处理0、6和24小时。裂解细胞做western blot分析。b为细胞处理同a,裂解细胞提取总RNA做FOXD3 qRT-PCR分析,actin作为内参 (n=3,***p<0.001)。c 1205Lu-TR HA-SOX10和A375-TR HA-SOX10细胞转染非靶向siRNA(对照)和SOX10特异性siRNA72小时,-/+100ng mL-1 Doxycycline处理72小时,然后2μM Vemurafenib处理细胞24小时,裂解细胞用来western blot分析。
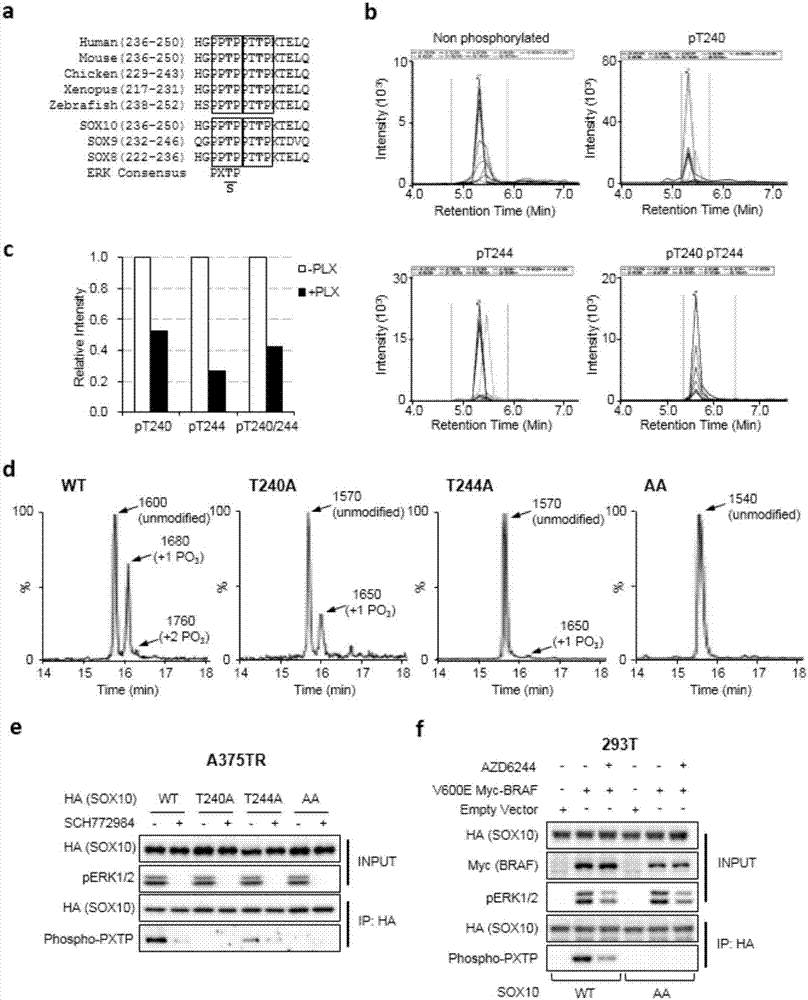
图2为ERK2可直接磷酸化SOX10 T240/T244位点。a为两个进化保守且在SOXE蛋白家族中保守的潜在ERK1/2激酶磷酸化基序,已在框中高亮显示,磷酸化位点(T)黑体加粗显示。b为体内检测SOX10T240/T244位点的磷酸化。A375-TR HA-SOX10细胞裂解液中免疫共沉淀HA-SOX10蛋白,胰酶酶切后多反应监测质谱技术分析。c为A375细胞给与溶剂对照或2 μM Vemurafenib处理6小时,对SOX10磷酸化质谱结果定量。SOX10随机酶切多肽片段(183AAQGEAECPGGEAEQGGTAAIQAHYK 208, 848.7188+++)作为内参。d为利用重组激活的ERK激酶和合成的SOX10多肽(236-HGPPTPPTTPKTELQ-250)进行体外激酶试验,反应产物进行LC-MS 分析。e为从慢病毒转染WT,T240A,T244A,AA HA-SOX10的A375细胞中免疫沉淀HA-SOX10蛋白,细胞给予溶剂对照或1μM SCH772984处理, anti-HA或者anti-phospho-PXTP检测蛋白样品。f在293T细胞中同时转染WT/AA HA-SOX10和空载/V600E BRAF质粒,给予溶剂对照或AZD6244处理。免疫沉淀HA-SOX10进行western blot检测。
图3为T240和(或)T244位点的磷酸化能抑制SOX10对FOXD3及其它下游靶基因的转录激活功能。a为在1205Lu-TR细胞中siRNA敲低内源 SOX10的同时外源过表达siRNA突变的HA-SOX10 T240E,T244E,and EE cDNA突变体,给予-/+100ng ml-1 doxycycline处理72小时。裂解细胞做 western blot分析,actin作为内参。b为A375-TR细胞给予处理同a。c为 1205Lu-TR或者A375-TR细胞感染WT或EE HA-SOX10慢病毒,同时siRNA 敲低内源SOX10,给予-/+100ng/ml doxycycline处理细胞72小时,然后给予 2μM Vemurafenib处理24h,裂解细胞提取总RNA,做qRT-PCR分析(n=3, *p<0.05;**p<0.01;***p<0.001)
图4为SOX10表达下调可以有效抑制FOXD3/ERBB3/AKT信号通路并显著促进抑制剂诱导的细胞凋亡。a黑色素瘤细胞转染对照siRNA或 SOX10#2 siRNA 72小时,±2μM vemurafenib处理24小时。随后10ng/ml NRG1刺激细胞1小时,裂解细胞做western blot分析。b 1205Lu-TR/A375-TR Ctrl-shRNA,SOX10 shRNA#1 and#2细胞给予100ng/ml doxycycline处理72 小时,将细胞重新悬浮后铺到96孔板,每孔5000个细胞,MTT试验监测 72小时细胞生长(n=3,*p<0.01;***p<0.001)。c黑色素瘤细胞转染对照 siRNA或SOX10#1、#2siRNA 48小时,±10μM vemurafenib处理48小时 (1205Lu细胞)或72小时(A375细胞)。随后收集细胞Annexin-V/PI染色,流式细胞仪检测分析(n=3,***p<0.001)。
图5为SOX10表达下调显著促进RAF抑制剂抑制肿瘤的效果并显著抑制肿瘤组织中FOXD3/ERBB3/AKT信号通路。a为建立感染对照shRNA或者SOX10-shRNA的1205Lu-TR or A375-TR细胞的裸鼠荷瘤模型,记录肿瘤生长曲线。(n=7,*p<0.05)。b为肿瘤记录第5天时,每组处死2只裸鼠,取肿瘤组织,做western blot分析。
具体实施方式
下面结合具体的实施例对本发明做进一步的详细说明,所述是对本发明的解释而不是限定。
下面提供本发明所需要的实验手段及实验操作。
1.细胞培养
携带BRAFV600E突变的黑色素瘤细胞株1205Lu、A375和M238以及 HEK293T细胞置于37℃恒温培养箱中培养(含5%CO2),其中A375、 HEK293T细胞在DMEM培养基中培养,1205Lu、M238细胞在RPMI 1640 培养基中培养。培养基均含10%的胎牛血清,青霉素(100单位/毫升)和链霉素(100微克/毫升)。
2.蛋白质印记
从细胞裂解液中提取总蛋白,在SDS-PAGE胶进行蛋白质分离,并印染至PVDF薄膜上。1%牛血清白蛋白封闭1小时后,用特定的一抗在4℃孵育过夜。第二天,用辣根过氧化物酶交联的二抗室温孵育1小时后,将条带在加强的化学发光仪上进行显影(BioRad,Hercules,CA,USA)。
Phospho-p44/42 MAPK(Thr202/Tyr204,clone 197G2,#4377)抗体, FOXD3(clone D20A9,#2019)抗体,HA-tag(clone 6E2,#2367,clone C29F4, #3724)抗体,Myc-tag(clone 71D10,#2278)抗体,HER3/ErbB3(clone 1B2E, #4754)抗体,Phospho-Akt(Ser473,clone D9E,#4060)抗体,AKT(#9272)抗体和 Phospho-MAPK Substrates Motif[PXpTP](#14378)抗体均购自Cell Signaling Technology(Beverley,MA,USA).β-actin(#A2066)抗体和FLAG-tag(clone M2,#F3165)购自Sigma-Aldrich(St.Louis,MO).SOX10(N-20,#SC-17342) 抗体购自Santa Cruz Biotechnology(Santa Cruz,CA,USA).另一个FOXD3 (#631702)抗体购自Biolegend(San Diego,CA,USA).
3.实时定量PCR
使用TriPure Isolation Reagent(Roche,Basel,Switzerland)从细胞中提取总 RNA,然后利用试剂盒(BioRad,Hercules,CA,USA)反转录成cDNA。使用iQ SYBR Green Supermix(BioRad)进行PCR反应,并利用CFX Connect real-time PCR detection system(BioRad)进行数据分析。
4.构建慢病毒包装载体和细胞系
野生型HA-SOX10 cDNA克隆至pENTR/D-TOPO vector(Thermo Fisher Scientific)来构成入门载体。随后入门载体与pLentipuro/TO/V5-DEST载体进行重组反应形成慢病毒载体。在HEK293FT细胞中进行慢病毒包装。将包装好的病毒感染黑色素瘤细胞72小时后,利用嘌呤霉素进行筛选。对于SOX10 shRNA质粒的构建是通过将DNA单链进行退火反应形成双链,随后连接至 pENTR/H1/TO质粒,构建成入门载体,随后重组至带有嘌呤霉素抗性的目的载体。同样,包装好的病毒来感染黑色素瘤细胞。
5.Annexin V/PI凋亡检测
收集细胞,用PBS清洗两次后根据试剂盒说明书染色20-30分钟 Annexin-V-FLUOS kit(Roche)。染色后的细胞用流式细胞仪进行检测 (Beckman Coulter,Indianapolis IN,USA)。利用Flowjo软件对数据进行分析 (Three Star,Inc.,Ashland,OR,USA)。
6.siRNA介导的靶基因抑制
待黑色素瘤细胞长至20-30%的融合度时,12.5nM小干扰RNA和转染试剂Lipofectamine RNAiMAX(Thermo Fisher Scientific)共同转染细胞72小时。非靶向的对照siRNA序列是(5′-UUCUCCGAACGUGUCACGU-3′),两条独立的SOX10siRNA序列分别是
#1 5′-CCGUAUGCAGCACAAGAAA-3′;
#2 5′-GUAUGCAGCACAAGAAAGA-3′。
以上siRNAs均购自上海吉玛公司(Shanghai,China)。
7.荷瘤小鼠实验
5周龄雌性BALB/c裸鼠购自上海斯莱克实验动物中心(Shanghai, China)。随机将裸鼠分成6组,每组8只。感染了Ctrl-shRNA,SOX10-shRNA #1或SOX10-shRNA#2的1205Lu-TR或A375-TR细胞皮间注射至裸鼠后臀部。分别注射1205Lu细胞2×106个/只或A375细胞4×106个每只。7-10天肿瘤长至较明显(40~100mm3),将裸鼠更换为添加了Doxycycline(2mg ml-1) 的饮水,并每日腹腔注射Vemurafenib或DMSO(30mg kg–1)。然后,每两天测一次肿瘤大小,肿瘤大小的计算公式为:体积=(长×宽2)×0.52。处理的第5天,每组处死两只裸鼠并取肿瘤,作为ERK/SOX10/FOXD3/ERBB3 信号通路的蛋白质印记分析。其余的老鼠继续记录数据并于第12天(A375) 或第14天(1206Lu)全部处死。以上所有动物实验都通过了西安交通大学动物保护和使用委员会的同意。
8.统计学分析
数据的显著性差异用单因素方差分析或双尾T检验进行分析。P值<0.05 时认为有显著性。Spearman’s相关性分析用来评估在黑色素瘤中基因SOX10 和FOXD3表达之间的相关性。
下面给出本发明实验的相关结果。
此前,Abel等人在黑色素瘤细胞中发现RAF抑制剂通过阻断ERK1/2 信号诱导转录因子FOXD3的表达,后者进而转录激活ERBB3的表达。ERBB3 的表达提高使肿瘤细胞对微环境中的生长因子Neuregulin 1(NRG1)敏感度增强,从而激活ERBB3及其下游的PI3K/AKT信号通路,增强肿瘤细胞对 RAF抑制剂的耐受。然而,RAF抑制剂如何诱导FOXD3表达,进而产生适应性耐药的关键调控分子尚未找到。在黑色素瘤中SOX10与FOXD3同样作为转录因子对神经脊的发育及维持胚胎干细胞全能性具有类似的作用。并且以往的ChIP-seq研究发现SOX10在FOXD3基因编码区的上游区域有结合位点。
在BRAF突变肿瘤病人组织中SOX10和FOXD3表达水平呈正相关
为验证SOX10的表达水平与FOXD3是否有相关性,我们利用生物信息学手段从TCGA数据库(http://cancergenome.nih.gov)获取BRAF突变黑色素瘤组织的RNA-seq数据,选取SOX10、FOXD3及其下游其他靶基因表达水平数据并进行Spearman相关性分析,观察它们表达水平之间是否存在正相关性。Spearman相关性分析结果不仅成功的检测到SOX10与其下游基因包括MITF,DCT和TYR存在显著正相关性,而且检测到SOX10与FOXD3表达水平呈显著正相关性(检测结果如图1所示)。所以,推测SOX10有可能是ERK1/2信号与下游FOXD3/ERBB3通路之间的桥梁分子,在ERBB3介导的适应性耐药中具有关键作用。
SOX10是RAF抑制剂诱导FOXD3表达的充分必要条件
为验证SOX10是ERK1/2信号下游调控FOXD3转录表达的充要因子,我们在多个黑色素瘤细胞系A375、1205Lu以及M238中发现SOX10的表达下调在蛋白水平(图2中的a所示)和RNA水平(图2中的b所示)都能有效阻断RAF抑制剂对FOXD3的诱导表达,提示SOX10是RAF抑制剂诱导FOXD3表达的必要条件。
为进一步检验SOX10是否为RAF抑制剂诱导FOXD3表达的充分条件,我们利用siRNA敲低内源SOX10的表达,利用慢病毒载体构建siRNA突变的过表达SOX10的细胞系,从而可以在敲低内源SOX10的蛋白水平而不影响外源SOX10的表达。观察表达外源SOX10在不受内源SOX10干扰的情况下能否有效恢复RAF抑制剂对FOXD3的表达诱导,从而验证SOX10对 FOXD3诱导表达的充分性。结果证明,外源过表达不仅可以增强RAF抑制剂诱导的FOXD3的表达,还可以在敲低内源SOX10的情况下几乎完全恢复FOXD3的诱导表达水平(图2中的c所示)。以上结果说明,SOX10是ERK1/2 信号下游调控FOXD3转录表达的充要因子。
ERK1/2激酶可以直接磷酸化SOX10的T240/T244位点
我们在SOX10蛋白序列中发现了两个进化保守的潜在ERK1/2激酶磷酸化基序(motif),同时也在SOXE蛋白家族中保守:ppTp(T240)和ptTp(T244) (图3中的a所示)。利用多反应监测质谱技术,我们验证了这两个位点在 BRAF突变黑色素瘤细胞中有磷酸化修饰,且其磷酸化水平在ERK信号被阻断后降低(图3中的b,c所示)。在此基础上,我们假设ERK1/2信号可能通过磷酸化SOX10T240/T244位点来抑制其对FOXD3基因的转录活性。
我们设计体外激酶实验来验证ERK激酶是否能直接磷酸化SOX10的 T240和T244位点。首先人工合成四种包含T240及T244位点的SOX10野生型及突变多肽片段,包括WT、T240A、T244A及AA SOX10片段。利用激活的重组ERK2激酶(购自NEB公司)及人工合成的SOX10多肽片段进行体外激酶实验。用LC-MS对激酶反应产物进行分析,检测野生型或突变 SOX10多肽被ERK2激酶磷酸化情况。结果显示,野生型(WT)多肽有三个峰,分别是非磷酸化多肽(分子量:1600D),单磷酸化多肽(分子量:1680D) 和双磷酸化多肽(分子量1760D)。T204A/T244A突变多肽只检测到非磷酸化峰和单个磷酸化峰。然而,在AA双突变的多肽并没有检测到任何磷酸化峰(图3中的d所示)。
为更进一步在体内证明ERK激酶可以磷酸化SOX10。我们在慢病毒感染的A375细胞中,给予对照或者ERK激酶抑制剂SCH772984处理,然后分别免疫共沉淀HA-SOX10 WT,T240A,T244A以及AA突变体,用可以靶向检测潜在ERK1/2激酶磷酸化基序的phospho-threonine抗体进行检测,结果证明在HA-SOX10 WT细胞中可以明显检测到phospho-threonine信号,在 SCH772984处理后信号显著下降。重要的是,在T240A,T244A以及AA突变体中都没有检测到phospho-threonine信号(图3中的e所示)。同样的,我们在293T细胞里也检测到SOX10 T240/T244位点的磷酸化信号,并且这种信号在给予RAF抑制剂后下降(图3中的f所示)。以上结果表明,ERK1/2 激酶可以直接磷酸化SOX10的T240/T244位点。
T240和(或)T244位点的磷酸化能抑制SOX10对FOXD3及其它下游靶基因的转录激活功能。
为验证SOX10磷酸化修饰对FOXD3及其它下游靶基因的转录激活功能的影响。我们构建了T240E、T244E以及EE SOX10等模拟磷酸化修饰突变的慢病毒载体及相应的黑色素瘤细胞株。用SOX10 siRNA敲低内源SOX10 的同时表达外源模拟磷酸化突变SOX10(T240E、T244E和EE),观察不同突变型SOX10对FOXD3转录表达的影响,并与野生型SOX10进行对比。同时观察SOX10磷酸化模拟突变对其转录激活其他目标基因(如TYR、MITF 和SAMMSON等)能力的影响,检验SOX10磷酸化修饰对其转录功能的抑制作用是否具有FOXD3特异性。
前面结果已显示,用SOX10 siRNA敲低内源SOX10的同时表达外源野生型SOX10可以几乎完全恢复RAF抑制剂对FOXD3的诱导表达(图2中的c所示)。但是,敲低内源SOX10同时外源过表达模拟磷酸化突变体SOX10 T240E/T244E可以一定程度抑制RAF抑制剂对FOXD3的诱导作用,并且EE 突变几乎完全抑制了FOXD3的诱导表达(图4中的a,b所示)。为研究SOX10 磷酸化对其它下游靶基因的转录激活功能的影响,我们将慢病毒感染 HA-SOX10 WT/EE的黑色素瘤细胞培养至25%-30%的融合度,一组加 Doxycycline诱导外源HA-SOX10的表达,另一组加溶剂对照。同时用RNAiMAX(invitrogen)转染试剂对两组细胞进行SOX10 siRNA或者随机序列siRNA的转染(对照)。72小时后用2uM的RAF抑制剂或DMSO处理细胞24小时,裂解细胞,用qRT-PCR分析SOX10的其他下游基因如MITF、SAMMSON以及TYR的表达情况。我们发现,磷酸化依赖的SOX10对其下游靶基因MITF、SAMMSON以及TYR的调控现象是普遍的。敲低内源SOX10可以降低这些靶基因的表达,再外源过表达野生型 SOX10可以完全恢复其表达水平,而SOX10 EE突变体却不能恢复(图4中的c所示)。以上结果有力的说明,ERK1/2可以通过磷酸化SOX10 T240/T244 位点抑制其对FOXD3及其它下游靶基因的转录活性。
SOX10表达下调可以有效抑制FOXD3/ERBB3/AKT信号通路并显著促进抑制剂诱导的细胞凋亡
在BRAF突变的黑色素瘤中,RAF抑制剂快速诱导FOXD3表达,上调 ERBB3并激活NRG1/ERBB3/AKT信号通路,进而产生适应性耐药。基于 SOX10对FOXD3/ERBB3通路的重要调控作用,我们推测敲低位于FOXD3 调控上游的SOX10蛋白,可以阻断FOXD3/ERBB3/AKT信号通路,并且增加黑色素瘤细胞对RAF抑制剂的敏感度。我们在A375和1205Lu黑色素瘤细胞中利用siRNA干扰技术下调SOX10蛋白的表达,利用western blot分析 SOX10蛋白下调情况,以及其下游FOXD3、ERBB3、phospho-ERBB3、AKT 和phospho-AKT等蛋白的表达情况。与之前的结果一致,RAF抑制剂可以增强ERBB3的表达并改善黑色素瘤细胞对ERBB3配体NRG1的敏感度。重要的是,敲低SOX10几乎完全阻断了RAF抑制剂诱导的ERBB3和依赖NRG1 激活的AKT信号的上调(图5中的a所示)。以上结果表明,SOX10可以通过调控NRG1/ERBB3/AKT信号通路的活性来调节FOXD3的表达。
我们设计了MTT实验和annexin V/PI staining实验来验证敲低SOX10对黑色素瘤细胞增殖和凋亡的影响。结果证明,单独敲低SOX10可以抑制黑色素瘤细胞增长,但是对细胞凋亡只有轻微的影响。然而,敲低SOX10的同时给予RAF抑制剂可以显著增加RAF抑制剂诱导的细胞凋亡。在A375细胞中,细胞凋亡从15%增加至27%,在1205Lu细胞中,从25%增加至48%(图5中的b,c所示)。结果表明,SOX10表达下调可以有效抑制 FOXD3/ERBB3/AKT信号通路并显著促进抑制剂诱导的细胞凋亡。
SOX10表达下调显著促进RAF抑制剂抑制肿瘤的效果并显著抑制肿瘤组织中FOXD3/ERBB3/AKT信号通路
我们将建好的黑色素瘤细胞(分别含SOX10-shRNA#1、#2和 control-shRNA)由皮间注射到4-6周大的免疫缺陷裸鼠背部(106细胞/只)。饲养至肿瘤生成并长大到可测量的大小。在所有小鼠的饮用水中添加200 ng/ml的Doxycycline(诱导shRNA表达),每周换水两次。通过腹腔注射方式给小鼠输送75mg/kg的RAF抑制剂(一天一次)或者溶剂对照(DMSO)。小鼠分为6组,每组8只。其中三组用对照处理,分别携带control-shRNA 和SOX10-shRNA#1、#2。另三组同样分别携带三种shRNA,用RAF抑制剂处理。每两天测量肿瘤大小一次(包括长和宽)并用如下公式计算肿瘤体积: 肿瘤体积=0.52x长度x宽度2。肿瘤生长至1000mm3时用CO2处死小鼠终止实验。收集裸鼠肿瘤组织用Western blot分析SOX10,FOXD3,ERBB3, phospho-ERBB3,AKT以及phospho-AKT等蛋白表达水平在对照组与处理组之间的差异。并绘制肿瘤生长曲线并对比control-shRNA组与SOX10-shRNA #1、#2组之间以及对照组与抑制剂组之间的差异。结果说明,在肿瘤组织中, SOX10敲低可以显著抑制RAF抑制剂对FOXD3/ERBB3/AKT信号的诱导表达,并且可以显著抑制肿瘤的生长。以上实验证明,SOX10敲低可以有效促进BRAF突变黑色素瘤对RAF抑制剂的敏感。
基于上述检测结果,本发明提出以下应用:
基因SOX10作为靶点在制备延缓黑色素瘤适应性耐药的药物中的应用。
进一步的:所述的药物是促使SOX10表达下调以抑制FOXD3的表达。
或者,所述的药物是使SOX10基因的T240和/或T244位点的磷酸化的药物,该药物能抑制FOXD3的转录活性。
或者,所述的药物是促使SOX10表达下调以促进RAF抑制剂抑制BRAF 突变黑色素瘤。
或者,所述的药物是促使SOX10表达下调以抑制FOXD3/ERBB3/AKT 信号通路并促进RAF抑制剂诱导的细胞凋亡。
或者,所述的药物是调节FOXD3表达的药物,SOX10通过调控NRG1/ ERBB3/AKT信号通路的活性来调节FOXD3的表达。
或者,所述的药物是延缓ERBB3介导的适应性耐药。
基于本发明所提供的基因SOX10可以作为靶点在制备延缓黑色素瘤适应性耐药的药物中进行应用,从而可以制备更好更多的延缓黑色素瘤适应性耐药的药物。
以上给出的实施例是实现本发明较优的例子,本发明不限于上述实施例。本领域的技术人员根据本发明技术方案的技术特征所做出的任何非本质的添加、替换,均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!