阿扎胞苷晶型的制备方法与流程
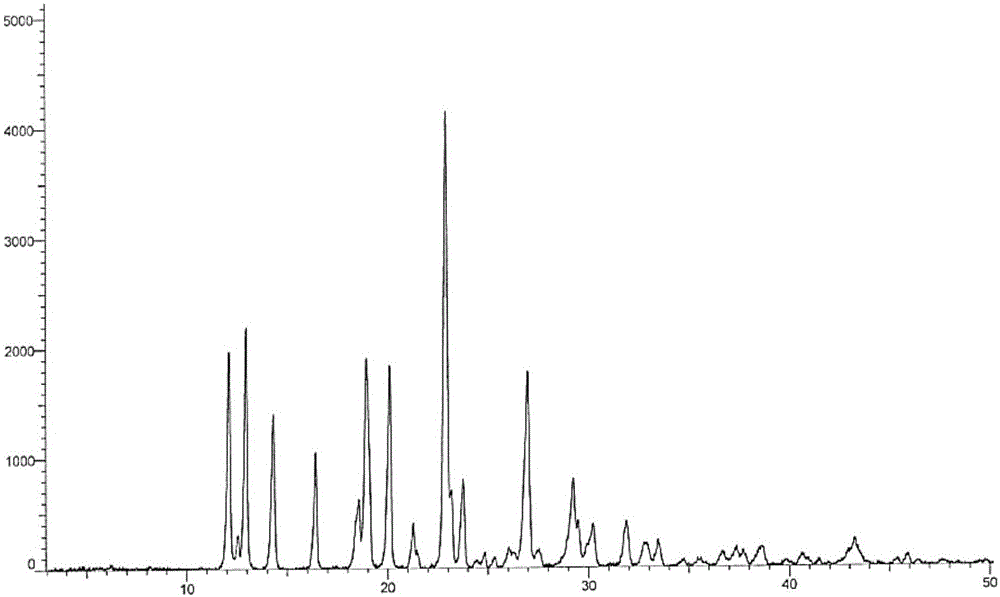
本发明涉及药物制备领域,具体涉及一种制备阿扎胞苷晶型i的方法。
背景技术:
:阿扎胞苷又名5-氮杂胞苷,化学名:1-(β-d-呋喃核糖基)-4-氨基-1,3,5-三嗪-2(1h)-酮。2004年5月阿扎胞苷成为美国fda批准上市的第一个用于治疗骨髓增生异常综合征(mds)的药物,2008年12月被欧盟批准上市,成为欧洲第一个可显著延长中级-2和高危mds和aml患者总生存期的治疗药物,2011年3月本药物在日本上市。阿扎胞苷在欧、美、日均享有罕见病用药地位。阿扎胞苷为胞苷的5-氮杂类似物,属于一类被称为低甲基化药物的表观遗传学抗肿瘤药。异常的dna甲基化使得调控正常细胞生长、分化和凋亡的关键基因失活,与肿瘤的发生和发展有关。阿扎胞苷治疗mds有效主要源自其dna低甲基化活性和对骨髓中异常造血细胞的直接细胞毒作用。阿扎胞苷为dna甲基转移酶抑制剂,其在最大抑制dna甲基化的浓度时并不会显著抑制dna合成。阿扎胞苷的结构式如式一所示:专利wo2004082619公开了阿扎胞苷8种晶型,分别是晶型i~ⅷ,其中最适合作为原料药的晶型为晶型i,其x-射线衍射特征峰为12.1、13.0、14.4、16.5、18.6、19.0、20.2、21.3、23.0、23.9、26.9、27.1、29.3、29.6、30.4、32.1,x-射线衍射图谱如图1所示,并且专利wo2004082822公开了阿扎胞苷晶型i的制备方法,即采用偶极非质子溶剂dmso、dmf、nmp等作为良溶剂,溶解阿扎胞苷,加入反溶剂改变溶液的极性,促使阿扎胞苷晶体析出。所用的反溶剂包括了醇类(特指c2-c5的醇)、酯类、醚类、烷烃、取代烷烃、酮类、腈类等7大类共计43种化学试剂。专利特别指出,采用异丙醇和乙腈能够稳定的得到晶型i。该专利几乎囊括了所有的常规化学试剂和常用实验方法。本发明人通过实验证实,采用专利wo2004082822提供的方法得到的产品存在着如下缺陷,第一:采用酯类、醚类、烷烃、取代烷烃、酮类溶剂作为反溶剂,得到的产品晶型为混晶,这给药品的质量控制带来困难;第二:采用乙腈作为反溶剂能够得到晶型i产品,但是溶剂残留问题严重,一般在1000ppm以上,药典规定乙腈限度为410ppm;第三:采用醇类作为反溶剂存在晶型纯度和溶剂残留的双重问题。综上,专利wo2004082822只是提供了制备阿扎胞苷晶型i的制备方法,但是在实际药物生产过程中采用这些方法无法生产出质量合格的药品。技术实现要素:针对上述缺陷,本发明进行了系统的研究,提供了一种制备低溶剂残留,高晶型纯度的阿扎胞苷晶型i的工艺。该工艺采用dmso作为溶剂,采用至少含有两种溶剂的混合溶剂作为反溶剂。混合溶剂由第一类溶剂和第二类溶剂组成,其中第一类溶剂为含碳原子数大于4的脂肪醇,优选碳原子数大于6的脂肪醇,特别优选碳原子数在6~8的脂肪醇;第二类溶剂为甲醇、乙醇以及含有甲醇、乙醇的溶剂。本发明提供了制备低溶剂残留,高晶型纯度的阿扎胞苷晶型i的操作方法:①在加热的条件下,用dmso溶解阿扎胞苷样品,②缓慢滴加反溶剂,③降温析晶,得到晶型i产品,其xrd谱图如附图1所示。根据本发明的制备工艺,加热温度选择大于40℃,优选大于60℃,最优选80~90℃。根据本发明的制备工艺,阿扎胞苷样品用适量的dmso溶解,加入反溶剂的体积应大于dmso体积的50%。根据本发明的制备工艺,反溶剂由第一类溶剂和第二类溶剂组成,第一类溶剂为碳原子数大于4的醇,优选c5~c8的醇,最优选直链c6~c7的醇,包括正己醇、正庚醇、正辛醇。第二类溶剂为甲醇、乙醇、含有甲醇、乙醇的有机溶剂,优选甲醇、乙醇,最优选甲醇;第二类溶剂所占混合溶剂的体积比例为30~80%,优选40%~70%,最优选50%~60%。根据本发明的制备工艺,加入反溶剂降温至40℃以下,优选0~40℃,更优选室温。根据本发明的制备工艺,样品进行真空干燥,干燥温度一般在60℃~120℃,优选70℃~100℃,最优选80℃~90℃。根据本发明制备方法制备的阿扎胞苷晶型i可用于治疗骨髓增生异常综合征。本发明对阿扎胞苷的溶解性进行了研究,测试了阿扎胞苷在下列溶剂中的溶解性:醇类溶剂,包括c1~c8的脂肪醇类如甲醇、乙醇、乙二醇、异丙醇、正丙醇、1,3-丙二醇、正丁醇、叔丁醇、正戊醇、2-戊醇、环戊醇、正己醇、环己醇、正庚醇、正辛醇等,和芳香醇类溶剂如苯甲醇;酯类溶剂包括甲酸乙酯、乙酸甲酯、乙酸乙酯、乙酸正丙酯、乙酸异丙酯、乙酸正丁酯,乙酸异丁酯、乙酸叔丁酯、乙酸正戊酯、乙酸异戊酯、丙酸甲酯、丙酸乙酯等;烷烃溶剂包括正戊烷、正己烷、环己烷、正庚烷、正辛烷等;取代烷烃溶剂包括二氯甲烷、氯仿、氯苯、硝基甲烷等;酮类溶剂包括丙酮、丁酮、2-丁酮,2-戊酮、环戊酮等;醚类溶剂包括乙醚、异丙醚、正丁醚、甲基叔丁基醚、苯甲醚、乙二醇甲醚、乙二醇二甲醚等;环醚类溶剂如四氢呋喃、1,4-二氧六环、2-甲基四氢呋喃等;含氰基溶剂如乙腈、丙腈、丁腈等。结果表明,阿扎胞苷在这些溶剂中均几乎不溶,这些溶剂不能作为析晶良溶剂,只适合作为不良溶剂。本研究还测试了20℃条件下阿扎胞苷在偶极非质子溶剂中的溶解性,溶剂包括二甲基亚砜(dmso),n,n-二甲基甲酰胺(dmf),n,n-二甲基乙酰胺(dma)和n-甲基吡咯烷酮(nmp),测试结果如下表:溶剂溶解度药典溶剂分类dmf0.04g/mliidma0.05g/mliinmp0.09g/mliidmso0.43g/mliii在溶解性方面,在dmso中溶解性是其他三种溶剂的4~10倍,在溶剂分类方面,dmf为ⅱ类溶剂,溶剂残留限度要求880ppm,dma为ⅱ类溶剂,溶剂残留限度要求为1090ppm,nmp为ⅱ溶剂,溶剂残留限度要求为530ppm,dmso为ⅲ类溶剂,溶剂残留限度要求为5000ppm。结果表明,dmso是阿扎胞苷析晶过程中理想的溶剂。本发明以dmso为溶剂,对不良溶剂进行了筛选,研究结果表明醚类、环醚类、酯类、烷烃类、取代烷烃类、酮类作为反溶剂结晶工艺很难控制,得到的样品晶型一般为多种晶型混合物,因此在阿扎胞苷原料药工业化生产上不能采用这些溶剂。本发明对含有氰基溶剂如乙腈、丙腈和丁腈进行了研究,发现这三种溶剂作为反溶剂均能稳定的得到晶型i样品,但是丙腈和丁腈在工业上基本不作为有机溶剂使用,采用乙腈生产,如上所述溶剂残留超标严重。本发明对醇类溶剂作为反溶剂的析晶方法进行了研究。研究内容包括产品的晶型纯度和产品的溶剂残留,研究结果表明,产品晶型纯度与采用的醇中含有的碳原子数目有很大关系,采用高级醇作为反溶剂,得到阿扎胞苷为纯的晶型i,采用低级醇作为反溶剂,得到的阿扎胞苷为晶型i与其他晶型的混合物,确切地说,当醇分子中碳原子数目大于2时,析晶得到的阿扎胞苷的晶型为晶型i,比如丙醇及碳原子数更多的醇;当醇分子碳原子数为1和2时,例如甲醇和乙醇,析晶得到的阿扎胞苷为晶型i与其他晶型的混合物;在另一方面,采用含有碳原子数大于2的高级醇作为反溶剂析晶能够得到纯的晶型i,但是存在一个弊端,即产品中dmso的残留量很高,本研究重点考察了从含有碳原子数1~8的醇作为反溶剂析晶的结果,采用甲醇作为反溶剂析晶时,样品残留dmso比较低,大约1000ppm,采用正辛醇作为反溶剂时,dmso的溶剂残留高达18000ppm,见附图7。针对晶型纯度和溶剂残留不能兼顾的情况,本研究创造性采用以两种及两种以上的不良溶剂作为反溶剂,第一种不良溶剂选择为c4~c8的脂肪醇,其作用为促使产生晶型i的样品,第二类不良溶剂的作用为降低样品中dmso的溶剂残留。通过大量试验筛查,确认类溶剂包括甲酸乙酯、乙酸甲酯、乙酸乙酯、乙酸正丙酯、乙酸异丙酯、乙酸正丁酯,乙酸异丁酯、乙酸叔丁酯、乙酸正戊酯、乙酸异戊酯、丙酸甲酯、丙酸乙酯等;烷烃溶剂包括正戊烷、正己烷、环己烷、正庚烷、正辛烷等;取代烷烃溶剂包括二氯甲烷、氯仿、氯苯、硝基甲烷等;酮类溶剂包括丙酮、丁酮、2-丁酮,2-戊酮、环戊酮等;醚类溶剂包括乙醚、异丙醚、正丁醚、甲基叔丁基醚、苯甲醚、乙二醇甲醚、乙二醇二甲醚等;环醚类溶剂如四氢呋喃、1,4-二氧六环、2-甲基四氢呋喃等不能有效的去除产品中的dmso溶剂残留;低级脂肪醇,如甲醇、乙醇等去除产品中dmso溶剂残留效果明显,但是加入量过多会导致产生混晶,异丙醇作为第二类反溶剂可以在一定程度上去除产品的dmso溶剂残留,但是产品中的dmso含量仍然很高,无法满足药典要求。以下是研究数据:表一:甲醇作为第二类溶剂的产品晶型情况表二:乙醇作为第二类溶剂的产品晶型情况表三:异丙醇作为第二类溶剂的产品晶型情况表四:甲醇作为第二类溶剂的产品dmso残留表五:乙醇作为第二类溶剂的产品dmso残留表六:异丙醇作为第二类溶剂的产品dmso残留附图说明图1:阿扎胞苷晶型i的xrd图谱。图2:阿扎胞苷晶型ⅲ的xrd图谱。图3:阿扎胞苷晶型ⅳ的xrd图谱。图4:阿扎胞苷晶型ⅰ和晶型ⅲ混合物的xrd图谱。图5:阿扎胞苷晶型ⅰ、ⅳ、ⅴ、ⅶ混合物的xrd图谱。图6:阿扎胞苷晶型ⅰ、ⅲ、ⅳ、ⅴ、ⅶ混合物的xrd图谱。图7:以不同醇溶剂作为反溶剂阿扎胞苷的溶剂残留量。具体实施方式下面结合附图和实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但并不用来限制本发明的范围。实施例1:将1g阿扎胞苷与5mldmso混合,加热至75℃,至样品全部溶解,缓慢加入由甲醇/正己醇=1:1组成的混合溶剂30ml,温度降至室温搅拌1h,过滤,样品在80℃真空干燥24h,获得0.8g晶型i样品,测得dmso的溶剂残留为830ppm,纯度大于99.9%,xrd图谱如图1所示。实施例2:将10g阿扎胞苷与40mldmso混合,加热至90℃,使样品溶解,缓慢加入由甲醇/正庚醇=1:1组成的混合溶剂200ml,温度降至室温后继续搅拌1h过滤,样品70℃干燥24h,获得晶型i样品9.1g,测得dmso溶剂残留1100ppm,纯度大于99.9%,xrd图谱与附图1基本一致。实施例3:将1g阿扎胞苷与8mldmso混合,加热至65℃使样品全部溶解,缓慢加入由乙醇/正辛醇=2:8组成的混合溶液80ml,温度降至室温继续搅拌1h过滤,在75℃下真空干燥20h得到晶型i样品0.88g,测得dmso溶剂残留5722ppm,xrd图谱与附图1基本一致。实施例4:将1g阿扎胞苷与6mldmso混合,加热至70℃使样品溶解,缓慢加入由甲醇/正辛醇=3:2组成的混合溶剂100ml,温度降至室温继续搅拌1h过滤,在70℃条件下真空干燥26h,获得晶型i样品0.84g,测得dmso溶剂残留1208ppm,纯度大于99.9%,xrd图谱与附图1基本一致。实施例5:将1g阿扎胞苷与7mldmso混合,加热至70℃使固体全部溶解,缓慢加入乙醇/正庚醇=2:8的混合溶剂40ml,温度降至室温继续搅拌1h过滤,样品70℃真空干燥22h,得到晶型i样品0.9g,测得dmso溶剂残留6423ppm,xrd图谱与附图1基本一致。实施例6:将1g阿扎胞苷与7mldmso混合,加热至75℃使固体溶解,缓慢加入甲醇/正己醇=4:1混合溶液40ml,冷却至室温,搅拌8h后过滤,得到的固体为晶型i和晶型ⅲ的混晶。xrd图谱如图4所示。实施例7:将1g阿扎胞苷与7mldmso混合,加热至75℃使固体溶解,缓慢加入甲醇40ml,冷却至室温,搅拌8h后过滤,得到的固体为晶型ⅰ、ⅳ、ⅴ、ⅶ的混晶。xrd图谱如图5所示。实施例8:将1g阿扎胞苷与7mldmso混合,加热至75℃使固体溶解,缓慢加入乙醇/正丁醇=9:1混合溶液40ml,冷却至室温,搅拌24h后过滤,得到的固体为晶型ⅰ、ⅲ、ⅳ、ⅴ、ⅶ的混晶。xrd图谱如图6所示。实施例9:将1g阿扎胞苷与7mldmso混合,加热至75℃使固体溶解,缓慢加入异丙醇45ml,冷却至室温搅拌3h过滤,在75℃条件下真空干燥24h,得到晶型i样品0.85g,测得溶剂残留6549ppm,xrd图谱与附图1基本一致。实施例10:将1g阿扎胞苷与7mldmso混合,加热至75℃使固体溶解,缓慢加入正己醇45ml,温度冷却至室温,继续搅拌3h过滤,75℃条件下真空干燥24h,得到晶型i样品0.87g,测得dmso溶剂残留16153ppm,xrd图谱与附图1基本一致。实施例11:将1g阿扎胞苷与7mldmso混合,加热至75℃使固体溶解,缓慢加入正辛醇45ml,温度冷却至室温,继续搅拌3h过滤,75℃条件下真空干燥24h,得到晶型i样品0.87g,测得dmso溶剂残留18376ppm,xrd图谱与附图1基本一致。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1