透气膜和制备所述透气膜的方法与流程
本发明涉及透气膜和制备所述透气膜的方法。
背景技术:
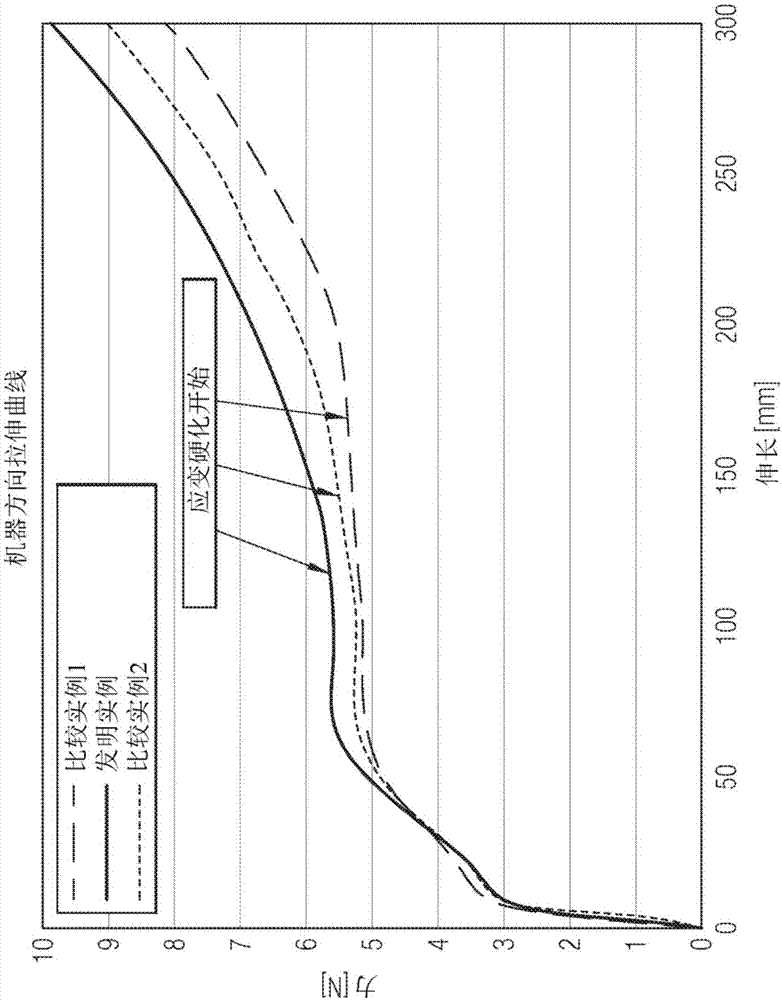
:通常已知在制造透气膜中使用聚乙烯组合物,如线性低密度聚乙烯。典型的工艺采用具有机器方向定向的流延膜挤出工艺以生产透气膜。在此类工艺中,挤出流延膜,并且然后其在机器方向上取向直至达到所需的基重(克/平方米(gramspersquaremeter,GSM))。由于应力局部化,难以在低取向水平下获得均匀的膜外观,这会产生膜缺陷,如虎纹(tigerstriping),即与较少变形区域条纹相邻的高度变形区域条纹。每种膜配方都具有固有的“最小拉伸比”,以达到所需的最终基重而不存在虎纹。仅在所述“最小拉伸比”之上,膜的光学外观是可接受的。然后,膜转换器微调拉伸比(在最小拉伸比之上)以达到机械性能的所需平衡。因此,具有低“最小拉伸比”的配方提供宽工艺窗口以及宽范围的最终膜性能,同时使膜缺陷最小化。尽管努力开发适用于透气膜生产应用的线性低密度聚乙烯组合物,但是仍然需要具有改进的机器方向定向,即低“最小拉伸比”的线性低密度聚乙烯组合物,同时提供改进的透气膜性能,如改进的水蒸气透过率、收缩率和穿刺强度。技术实现要素:本公开提供透气膜和制备所述透气膜的方法。在一个实施例中,本公开提供透气膜,其包含膜层,所述膜层包含聚合物组合物,所述聚合物组合物包含等于或小于60重量%的线性低密度聚乙烯树脂,其表现出以下各种性质:(1)70至90℃的CEF级分等于或大于总CEF级分的80%;(2)根据ASTMD1238(2.16kg在190℃下)测量的熔融指数(meltindex)I2在等于或大于2.0克/10分钟且等于或小于5.0克/10分钟的范围内;和(3)熔体流动比(meltflowratio)I10/I2等于或小于6.7。在一个替代实施例中,本公开进一步提供一种制备透气膜的方法,其包含:(a)流延挤出聚合物组合物,其包含大于0重量%至60重量%的线性低密度聚乙烯,其表现出以下各种性质:(1)70至90℃的CEF级分等于或大于总CEF级分的80%;(2)根据ASTMD1238(2.16kg在190℃下)测量的熔融指数(meltindex)I2等于或大于2.0克/10分钟且等于或小于5克/10分钟;和(3)熔体流动比I10/I2等于或小于6.7,以制备流延挤出膜层;和(b)机器方向定向所述流延挤出膜层。在一个替代实施例中,本公开进一步提供一种制备透气膜的方法,其包含:(a)吹塑挤出聚合物组合物,其包含大于0重量%至60重量%的线性低密度聚乙烯,其表现出以下各种性质:(1)70至90℃的CEF级分等于或大于总CEF级分的80%;(2)根据ASTMD1238(2.16kg在190℃下)测量的熔融指数I2等于或大于2.0克/10分钟且等于或小于5.0克/10分钟;和(3)熔体流动比I10/I2等于或小于6.7,以制备吹塑膜层;和(b)机器方向定向吹塑膜层。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了线性低密度聚乙烯树脂进一步表现出0.915至0.940g/cm2的密度(根据ASTMD792测量)之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的制备方法,除了线性低密度聚乙烯包含衍生自乙烯的单元和衍生自一种或多种α-烯烃共聚单体的单元之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了共聚单体是1-己烯之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了共聚单体是1-辛烯之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了共聚单体是1-丁烯之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了线性低密度聚乙烯树脂不包含衍生自辛烯的单元之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了聚合物组合物进一步包含40至60重量%的CaCO3之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了聚合物组合物包含30至60重量%的线性低密度聚乙烯树脂之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了聚合物组合物进一步包含大于0至等于或小于5重量%的一种或多种选自由颜料和抗氧化剂组成的组的化合物之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了聚合物组合物进一步包含大于0至等于或小于10重量%的一种或多种选自由聚丙烯和低密度聚乙烯组成的组的化合物之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了至少一个膜层包含聚合物组合物之外,所述聚合物组合物包含30至60重量%的线性低密度聚乙烯树脂,其表现出以下各种性质:(1)70至90℃的CEF级分等于或大于总CEF级分的80%;(2)根据ASTMD1238(2.16kg在190℃下)测量的熔融指数I2等于或大于2.0克/10分钟且等于或小于5.0克/10分钟;和(3)熔体流动比I10/I2等于或小于6.7。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了至少一个薄膜层包含聚合物组合物之外,所述聚合物组合物包含(a)45至55重量%的聚线性低密度聚乙烯树脂,其表现出以下各种性质:(1)70至90℃的CEF级分等于或大于总CEF级分的80%;(2)根据ASTMD1238(2.16kg在190℃下)测量的熔融指数I2等于或大于2.0克/10分钟且等于或小于5.0克/10分钟;和(3)熔体流动比I10/I2等于或小于6.7;和(b)55至45重量%的CaCO3。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了机器方向定向以等于或大于1.5的拉伸比进行之外。在一个替代实施例中,本公开进一步提供根据前述实施例中任一项的透气膜和制备所述透气膜的方法,除了透气膜包括一个或多个附加膜层之外。附图说明出于说明本发明的目的,在附图中示出示例性的形式;然而,应理解,本发明不限于所示的精确装置和仪器。图1是说明根据ISO527-3测量的由发明和比较组合物形成的膜在机器方向上的拉伸曲线特性的图;图2是说明发明和比较组合物的CEF分析的图;图3是说明由发明和比较复合组合物形成的膜的抗穿刺性的图;图4是说明由发明和比较复合组合物形成的膜的水头(hydrohead)的图;图5是说明由发明和比较复合组合物形成的膜的水蒸汽透过率(38℃-90%RH)性质的图;图6是说明由发明和比较复合组合物形成的膜在5%伸长率下在机器方向上的拉伸强度性质的图;并且图7是说明发明和比较实例在机器方向上的收缩性质的图(浸没在80℃的水浴中达30秒)。具体实施方式本公开提供透气膜和制备所述透气膜的方法。根据本公开的所述透气膜包含膜层,所述膜层包含聚合物组合物,所述聚合物组合物包含等于或小于60重量%的线性低密度聚乙烯树脂,其表现出以下各项性能:(1)70至90℃的CEF级分等于或大于总CEF级分的80%;(2)根据ASTMD1238(2.16kg在190℃下)测量的熔融指数I2在等于或大于2.0克/10分钟且等于或小于5.0克/10分钟的范围内;和(3)熔体流动比I10/I2等于或小于6.7。根据本公开的制备透气膜的方法包含:(a)流延挤出聚合物组合物,其包含大于0重量%至60重量%的线性低密度聚乙烯,其表现出以下各种性质:(1)70至90℃的CEF级分等于或大于总CEF级分的80%;(2)根据ASTMD1238(2.16kg在190℃下)测量的熔融指数I2等于或大于2.0克/10分钟且等于或小于5.0克/10分钟;并且(3)熔体流动比I10/I2等于或小于6.7,以制备流延挤出膜层;和(b)机器方向定向所述流延挤出膜层。在一个替代实施例中,根据本公开的制备透气膜的方法包含:(a)吹塑挤出聚合物组合物,其包含大于0重量%至60重量%的线性低密度聚乙烯,其表现出以下各种性质:(1)70至90℃的CEF级分等于或大于总CEF级分的80%;(2)根据ASTMD1238(2.16kg在190℃下)测量的熔融指数I2等于或大于2.0克/10分钟且等于或小于5.0克/10分钟;和(3)熔体流动比I10/I2等于或小于6.7,以制备吹塑膜层;和(b)机器方向定向吹塑膜层。以聚合物组合物的重量计,聚合物组合物包含60重量%或更少的线性低密度聚乙烯,下面进一步描述,例如,以聚合物组合物的重量计,30至60重量%的线性低密度聚乙烯。以聚合物组合物的重量计,聚合物组合物包含40至60重量%的CaCO3。以聚合物组合物的重量计,聚合物组合物可进一步包含10重量%或更少的低密度聚乙烯或聚丙烯。聚合物组合物可进一步包含额外组分,如一种或多种添加剂。此类添加剂包括但不限于抗静电剂、增色剂、染料、润滑剂、填充剂(如TiO2)、遮光剂、成核剂、加工助剂、颜料、主抗氧化剂、次抗氧化剂、加工助剂、UV稳定剂、抗阻断剂、增滑剂、增粘剂、阻燃剂、抗微生物剂、减臭剂、抗真菌剂和其组合。以包括此类添加剂的聚合物组合物的重量计,聚合物组合物可含有约0.1至约10%组合重量的此类添加剂。线性低密度聚乙烯(LinearLowDensityPolyethylene,LLDPE)线性低密度聚乙烯(LLDPE)表现出以下各种性质:(1)70至90℃的CEF级分等于或大于总CEF级分的80%;(2)根据ASTMD1238(2.16kg在190℃下)测量的熔融指数I2在等于或大于2.0克/10分钟且等于或小于5.0克/10分钟的范围内;和(3)熔体流动比I10/I2等于或小于6.7。线性低密度聚乙烯(LLDPE)包含乙烯/α-烯烃共聚物,其包含(a)小于或等于60重量%,例如至少70重量%,或至少80重量%,或至少90重量%的衍生自乙烯的单元;和(b)小于30重量%,例如小于25重量%,或小于20重量%,或小于10重量%的衍生自一种或多种α-烯烃共聚单体的单元。术语“乙烯/α-烯烃共聚单体”是指含有大于50摩尔%聚合乙烯单体(以可聚合单体的总量计)和至少一种其它共聚单体的聚合物。在一个具体实施例中,乙烯/α-烯烃共聚物具有衍生自乙烯的单元和衍生自两种不同α-烯烃共聚单体的单元。通常α-烯烃共聚单体的碳原子不超过20个。例如,优选地α-烯烃共聚单体可具有3至8个碳原子,并且更优选地3至6个碳原子。示例性α-烯烃共聚单体包括但不限于丙烯、1-丁烯、1-戊烯、1-己烯、1-庚烯、1-辛烯、1-壬烯、1-癸烯和4-甲基-1-戊烯。一种或多种α-烯烃共聚单体可例如选自由丙烯、1-丁烯、1-己烯组成的组,或替代地,选自由1-丁烯和1-己烯组成的组。在一个实施例中,线性低密度聚乙烯树脂不包含衍生自1-辛烯的单元。根据ASTMD1238(2.16kg在190℃下)测量的LLDPE的熔融指数I2在等于或大于2.0克/10分钟且等于或小于5.0克/10分钟的范围内。本文包括并公开2.0至5.0克/10分钟的所有单个值和子范围;例如,I2可在2.0、3.0或4.0克/10分钟的下限至3.0、4.0或5.0克/10分钟的上限范围内。例如,I2可为2.0至5.0克/10分钟,或替代地,2.0至3.5克/10分钟,或替代地,3.5至5.0克/10分钟,或替代地,3.0至4.0克/10分钟。LLDPE的特征在于零剪切粘度比(zeroshearviscosityratio,ZSVR)在1.2至5.0的范围内。本文包括并公开1.2至5.0范围内的所单个值和子范围;例如,ZSVR可以在1.2、1.4、1.6、1.8的下限至2.0、3.0、4.0或5.0的上限范围内。例如,ZSVR可在1.2至5.0,或替代地,1.5至4,或替代地,1.8至3.5的范围内。LLDPE的密度在0.915至0.940g/cm3,例如0.915至0.925g/cm3的范围内。本文包含并公开0.915至0.940g/cm3的所有单个值和子范围;例如,密度可以在0.915、0.920、0.925、0.930或0.935g/cm3的下限至0.917、0.922、0.927、0.932、0.937或0.940g/cm3的上限范围内。例如,密度可以为0.915至0.940g/cm3,或替代地,0.915至0.927g/cm3,或替代地,0.927至0.940g/cm3,或替代地,0.915至0.921g/cm3。LLDPE的分子量分布(Mw/Mn)在2.0至3.5的范围内。本文包括并公开2.0至3.5的所有单个值和子范围;例如,分子量分布(Mw/Mn)可以在2、2.1、2.2、2.4、2.5或2.6的下限至2.2、2.3、2.4、2.5、2.7、2.9、3.2或3.5的上限范围内。例如,分子量分布(Mw/Mn)可以为2.0至3.5,或替代地,2.0至2.4,或替代地,2.0至2.8,或替代地,2.8至3.5。LLDPE的分子量分布(Mz/Mn)在3.5至6的范围内。本文包括并公开3.5至6的所有单个值和子范围;例如,分子量分布(Mz/Mn)可以为3.5、3.7、3.9、4.5或5的下限至3.5、4.0、4.2、4.4、4.7、5.0、5.5或6.0的上限。例如,分子量分布(Mz/Mn)可以在3.5至6,或替代地,可以是3.5至4.8,或替代地,4.8至6,或替代地,4至5,或替代地,3.5至4.5的范围内。LLDPE的分子量分布不对称性[(Mw/Mn)/(Mz/Mw)]即Mw2/(Mn*Mz)在1.00至1.40的范围内。例如,分子量分布不对称性Mw2/(Mn*Mz)可以为1.0、1.05、1.10、1.15或1.20的下限至1.25、1.30、1.35或1.40的上限。例如,分子量分布不对称性Mw2/(Mn*Mz)可以在1.00至1.40,或替代地,从1.00至1.20,或替代地,从1.20至1.40,或替代地,从1.10至1.30的范围内。LLDPE的乙烯基不饱和度为LLDPE的主链中存在的每百万个碳原子中的乙烯基少于150个。本文包括并公开每百万个碳原子中的乙烯基少于150个的所有单个值和子范围;例如,乙烯基不饱和度可以为LLDPE的主链中存在的每百万个碳原子中的乙烯基少于150个,或替代地,少于120个,或替代地,少于80个,或替代地,少于50个。LLDPE的结晶热在135至145J/g的范围内。本文包括并公开135至145J/g的所有单个值和子范围;例如,结晶热可以为135、136、137或138J/g的下限至140、141、143或145J/g的上限。例如,结晶热可以在135至145J/g,或替代地,135至140J/g,或替代地,140至145J/g,或替代地,137至142J/g的范围内。LLDPE的峰值结晶温度在94至101℃的范围内。本文包括并公开94至101℃的所有单个值和子范围;例如,峰值结晶温度可以为94、95、96或97℃的下限至98、99、100或101℃的上限。例如,峰值结晶温度可以为94至101℃,或替代地,94至97℃,或替代地,97至101℃,或替代地,95至99℃。LLDPE的熔化热在135至145J/g的范围内。本文包括并公开135至145J/g的所有单个值和子范围;例如,熔化热可以为135、136、137或138J/g的下限至140、141、143或145J/g的上限。例如,熔化热可以为135至145J/g,或替代地,135至140J/g,或替代地,140至145J/g,或替代地,137至142J/g。LLDPE的峰值熔化温度在108至116℃的范围内。本文包括并公开94至101℃的所有单个值和子范围;例如,峰值熔化温度可以为108、109、110或11℃的下限到113、114、115或116℃的上限。例如,峰值熔化温度可以为108至116℃,或替代地,108至112℃,或替代地,112至116℃,或替代地,110至114℃。在一个实施例中,每百万重量份的LLDPE,LLDPE包含小于或等于100重量份,例如小于10重量份,小于8重量份,小于5重量份,小于4重量份,小于1重量份,小于0.5重量份或小于0.1重量份的包含多价芳氧基醚的金属络合物的催化剂体系剩余的金属络合物残余物。LLDPE中的包含多价芳氧基醚的金属络合物的催化剂体系剩余的金属络合物残余物可通过x射线荧光(x-rayfluorescence,XRF)测量,其根据参考标样校准。在优选的方法中,可以将聚合物树脂颗粒在升高的温度下压缩模制成约3/8英寸厚的板,用于x射线测量。在非常低的金属络合物浓度下,如低于0.1ppm,ICP-AES将是测定存在于LLDPE中的金属络合物残余物的适合方法。LLDPE可进一步包含额外组分,如一种或多种其它聚合物和/或一种或多种添加剂。此类添加剂包括(但不限于)抗静电剂、增色剂、染料、润滑剂、填充剂(如TiO2或CaCO3)、遮光剂、成核剂、加工助剂、颜料、主抗氧化剂、次抗氧化剂、加工助剂、UV稳定剂、抗阻断剂、增滑剂、增粘剂、阻燃剂、抗微生物剂、减臭剂、抗真菌剂和其组合。以包括此类添加剂的LLDPE的重量计,LLDPE可含有约0.1%至约10%组合重量的此类添加剂。可使用任何常规的乙烯(共)聚合溶液单反应器反应工艺来制备LLDPE。制备本文公开的LLDPE的一种方法详细描述于美国专利5,977,251中,所述专利的公开内容以整体引用的方式并入本文。在一个实施例中,LLDPE经由聚合工艺在单溶液相环路反应器系统中制备,其中催化剂体系包含(a)一种或多种前催化剂,其包含下式(I)的金属配体络合物:M是钛、锆或铪,各自独立地处于+2、+3或+4形式氧化态;并且n为0至3的整数,并且其中当n为0时,X不存在;并且每个X独立地为中性、单阴离子或双阴离子单齿配体;或两个X连在一起形成中性、单阴离子或双阴离子双齿配体;并且以使式(I)的金属配体络合物总体上是中性的方式选择X和n;并且每个Z独立地为O、S、N(C1-C40)烃基或P(C1-C40)烃基;L为(C2-C40)亚烃基或(C2-C40)杂亚烃基,其中(C2-C40)亚烃基具有包含连接式(I)中Z原子(L与其键合)的2-碳原子至10-碳原子连接基团主链的部分,并且(C2-C40)杂亚烃基具有包含连接式(I)中Z原子的3-原子至10-原子连接基团主链的部分,其中(C2-C40)杂烃基的3-原子至10-原子连接基团主链的3至10个原子中的每一个独立地为碳原子或杂原子,其中每个杂原子独立地为O、S、S(O)、S(O)2、Si(RC)2、Ge(RC)2、P(RP)或N(RN),其中每个RC独立地选自由(C1-C40)烃基组成的组。如本文所用,RC包括其中两个RC基团连接在一起形成双自由基环并且其中Si在环内的情况。每个RP是(C1-C40)烃基;并且每个RN是(C1-C40)烃基或不存在;并且R1-10各自独立地选自由以下组成的组:(C1-C40)烃基、(C1-C40)杂烃基、Si(RC)3、Ge(RC)3、P(RP)2、N(RN)2、ORC、SRC、NO2、CN、CF3、RCS(O)-、RCS(O)2-、(RC)2C=N-、RCC(O)O-、RCOC(O)-、RCC(O)N(R)-、(RC)2NC(O)-、卤素原子、氢原子和其任何组合,并且至少两个Y1-Y3和至少两个Y4-Y6是氟原子,并且当仅两个Y1-Y3和仅两个Y4-Y6是氟原子时,非氟Y1-Y6选自由H原子、烷基、芳基、杂芳基和烷氧基组成的组,并且任选地,R1-10基团的两个或更多个R基团(例如,来自R1-4、R5-8)可以结合在一起成环结构,此类环结构,在环中具有2至50个原子,不包括任何氢原子。如本文所用,术语“(Cx-Cy)烃基”意指x至y个碳原子的烃自由基,并且术语“(Cx-Cy)亚烃基”意指x至y个碳原子的烃双自由基,并且术语“(Cx-Cy)烷基”意指x至y个碳原子的烷基,并且术语“(Cx-Cy)环烷基”意指x至y个碳原子的环烷基。如本文所用,术语“(C1-C40)烃基”意指1至40个碳原子的烃自由基,并且术语“(C2-C40)亚烃基”意指2至40个碳原子的烃双自由基,其中每个烃自由基和双自由基独立地为芳香族(6个碳原子或更多)或非芳香族、饱和或不饱和、直链或支链、环状(包括单环和多环、稠合和非稠合多环,包括双环;3个碳原子或更多个)或非环状的,或其两种或多种的组合;并且每个烃自由基和双自由基各自独立地与另一个烃自由基和双自由基相同或不同,并且独立地为未取代的或由一个或多个RS取代。优选地,(C1-C40)烃基独立地为未取代或取代(C1-C40)烷基、(C3-C40)环烷基、(C3-C20)环烷基-(C1-C20)亚烷基、(C6-C40)芳基或(C6-C20)芳基-(C1-C20)亚烷基。更优选地,前述(C1-C40)烃基中的每一个独立地具有最多20个碳原子(即(C1-C20)烃基),并且仍更优选地最多12个碳原子。术语“(C1-C40)烷基”和“(C1-C18)烷基”分别意指1至40个碳原子或1至18个碳原子的饱和直链或支链烃自由基,其为未取代的或由一个或多个Rs取代。未取代(C1-C40)烷基的实例是未取代(C1-C20)烷基;未取代(C1-C10)烷基;未取代(C1-C5)烷基;甲基;乙基;1-丙基;2-丙基;1-丁基;2-丁基;2-甲基丙基;1,1-二甲基乙基;1-戊基;1-己基;1-庚基;1-壬基和1-癸基。取代(C1-C40)烷基的实例是取代(C1-C20)烷基、取代(C1-C10)烷基、三氟甲基和(C45)烷基。(C45)烷基是,例如,由一个Rs取代的(C27-C40)烷基,其分别是(C18-C5)烷基。优选地,每个(C1-C5)烷基独立地为甲基、三氟甲基、乙基、1-丙基、1-甲基乙基或1,1-二甲基乙基。术语“(C6-C40)芳基”意指6至40个碳原子的未取代或取代(由一个或多个RS)单-、双-或三环芳香族烃自由基,其中至少6至14个碳原子是芳香环碳原子,并且单-、双-或三环自由基分别包含1个、2个或3个环;其中1个环是芳香族的,并且2个或3个环独立地稠合或非稠合,并且2个或3个环中的至少一个是芳香族的。未取代(C6-C40)芳基的实例是未取代(C6-C20)芳基;未取代(C6-C18)芳基;2-(C1-C5)烷基-苯基;2,4-双(C1-C5)烷基-苯基;苯基;芴基;四氢芴基;引达省基(indacenyl);四氢引达省基(hexahydroindacenyl);茚基;二氢茚基;萘基;四氢萘基;和菲。取代(C6-C40)芳基的实例是取代(C6-C20)芳基;取代(C6-C18)芳基;2,4-双[(C20)烷基]-苯基;聚氟苯基;五氟苯基;和芴-9-酮-1-基。术语“(C3-C40)环烷基”意指3至40个碳原子的饱和环烃自由基,其为未取代的或由一个或多个Rs取代。其它环烷基(例如,(C3-C12)环烷基))以类似方式定义。未取代(C3-C40)环烷基的实例是未取代(C3-C20)环烷基、未取代(C3-C10)环烷基、环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基和环癸基。取代(C3-C40)环烷基的实例是取代(C3-C20)环烷基、取代(C3-C10)环烷基、环戊烷-2-基和1-氟环己基。(C1-C40)亚烃基的实例是未取代或取代(C6-C40)亚芳基、(C3-C40)亚环烷基和(C1-C40)亚烷基(例如,(C1-C20)亚烷基)。在一些实施例中,双自由基在相同的碳原子(例如,-CH2-)上或在相邻的碳原子上(即1,2-双自由基),或由一个、两个或更多个介入式碳原子间隔开(例如,相应的1,3-双自由基、1,4-双自由基等)。优选1,2-、1,3-、1,4-或α,ω-双自由基,并且更优选地1,2-双自由基。α,ω-双自由基是自由基碳之间碳主链间距最大的双自由基。更优选1,2-双自由基、1,3-双自由基或1,4-双自由基形式的(C6-C18)亚芳基、(C3-C20)亚环烷基或(C2-C20)亚烷基。术语“(C3-C40)亚环烷基”意指3至40个碳原子的环双自由基(即,自由基在环原子上),其为未取代的或由一个或多个Rs取代。未取代(C3-C40)亚环烷基的实例是1,3-亚环丙基、1,1-亚环丙基和1,2-亚环己基。取代(C3-C40)亚环烷基的实例是2-氧代-1,3-亚环丙基和1,2-二甲基-1,2-亚环己基。术语“(C1-C40)杂烃基”意指1至40个碳原子的杂烃自由基,并且术语“(C1-C40)杂亚烃基”意指1至40个碳原子的杂烃双自由基,并且每个杂烃独立地具有一个或多个杂原子O;S;S(O);S(O)2;Si(RC)2;Ge(RC)2;P(RP)和N(RN),其中每个RC独立地为未取代(C1-C40)烃基,每个RP为未取代(C1-C40)烃基;并且每个RN为未取代(C1-C40)烃基或不存在(例如,当N包含-N=或三碳取代N时不存在)。杂烃自由基和每个杂烃双自由基独立地在碳原子或其杂原子上,但优选地当与式(I)中的杂原子键合或与另一个杂烃基或杂亚烃基的杂原子键合时在碳原子上。每个(C1-C40)杂烃基和(C1-C40)杂亚烃基独立地为未取代或取代的(由一个或多个Rs)、芳香族或非芳香族的、饱和或不饱和的、直链或支链、环状(包括单环和多环、稠合和非稠合多环)或非环状的,或其两种或多种的组合;并且每一个各自与另一个相同或不同。优选地,(C1-C40)杂烃基独立地为未取代或取代(C1-C40)杂烷基、(C1-C40)烃基-O-、(C1-C40)烃基-S-、(C1-C40)烃基-S(O)-、(C1-C40)烃基--S(O)2-、(C1-C40)烃基-Si(RC)2-、(C1-C40)烃基-Ge(RC)2-、(C1-C40))烃基-N(RN)-、(C1-C40)烃基-P(RP)-、(C2-C40)杂环烷基、(C2-C19)杂环烷基-(C1-C20)亚烷基、(C3-C20)环烷基-(C1-C19)杂亚烷基、(C2-C19)杂环烷基-(C1-C20)杂亚烷基、(C1-C40)杂芳基、(C1-C19)杂芳基-(C1-C20)亚烷基、(C6-C20)芳基-(C1-C19)杂亚烷基或(C1-C19)杂芳基-(C1-C20)杂亚烷基。术语“(C1-C40)杂芳基”意指1至40个总碳原子和1至4个杂原子的未取代或取代(由一个或多个RS)单-、双-或三环杂芳香烃自由基,并且单-、双环或三环自由基分别包含1个、2个或3个环,其中2个或3个环独立地稠合或非稠合,并且2个或3个环中的至少一个是杂芳香族的。其它杂芳基(例如,(C4-C12)杂芳基))以类似方式定义。单环杂芳香烃自由基为5元或6元环。5元环分别具有1至4个碳原子和4至1个杂原子,每个杂原子为O、S、N或P,并且优选地O、S或N。5元环杂芳香烃自由基的实例是吡咯-1-基;吡咯-2-基;呋喃-3-基;噻吩-2-基;吡唑-1-基;异恶唑-2-基;异噻唑-5-基;咪唑-2-基;恶唑-4-基;噻唑-2-基;1,2,4-三唑-1-基;1,3,4-恶二唑-2-基;1,3,4-噻二唑-2-基;四唑-1-基;四唑-2-基;和四唑-5-基。6元环具有4或5个碳原子和2或1个杂原子,杂原子为N或P,并且优选地N。6元环杂香烃自由基的实例是吡啶-2-基;嘧啶-2-基;和吡嗪-2-基。双环杂芳烃基优选为稠合的5,6-或6,6-环系。稠合5,6-环体系双环杂芳香烃自由基的实例是吲哚-1-基;和苯并咪唑-1-基。稠合6,6-环体系双环杂芳香烃自由基的实例是喹啉-2-基;和异喹啉-1-基。优选地三环杂芳香烃自由基为稠合5,6,5-;5,6,6-;6,5,6-;或6,6,6-环体系。稠合5,6,5-环体系的实例是1,7-二氢吡咯并[3,2-f]吲哚-1-基。稠合5,6,6-环体系的实例是1H-苯并[f]吲哚-1-基。稠合6,5,6-环体系的实例是9H-咔唑-9-基。稠合6,6,6-环体系的实例是吖啶-9-基。在一些实施例中,(C1-C40)杂芳基是2,7-二取代咔唑基或3,6-二取代咔唑基或未取代咔唑,更优选地其中每个Rs独立地为苯基、甲基、乙基、异丙基或叔丁基,仍更优选地2,7-二(叔丁基)-咔唑基、3,6-二(叔丁基)-咔唑基、2,7-二(叔辛基)-咔唑基、3,6-二(叔辛基)-咔唑基、2,7-二苯基咔唑基、3,6-二苯基咔唑基、2,7-双(2,4,6-三甲基苯基)-咔唑基或3,6-双(2,4,6-三甲基苯基)-咔唑基。前述杂烷基和杂亚烷基分别为饱和直链或支链自由基或双自由基,含有(C1-C40)碳原子,或根据具体情况含有较少的碳原子,和一个或多个如上定义的杂原子Si(RC)2、Ge(RC)2、P(RP)、N(RN)、N、O、S、S(O)和S(O)2,其中杂烷基和杂亚烷基中的每一个独立地为未取代的或由一个或多个RS取代。未取代(C2-C40)杂环烷基的实例是未取代(C2-C20)杂环烷基、未取代(C2-C10)杂环烷基、氮杂环丙烷-1-基、氧杂环丁烷-2-基,四氢呋喃-3-基、吡咯烷-1-基、四氢噻吩-S,S-二氧化物-2-基、吗啉-4-基、1,4-二恶烷-2-基、六氢氮杂卓-4-基、3-氧杂-环辛基、5-硫代-环壬基和2-氮杂-环癸基。术语“卤素原子”意指氟原子(F)、氯原子(Cl)、溴原子(Br)或碘原子(I)自由基。优选地每个卤素原子独立地为Br、F或Cl自由基,并且更优选地F或Cl自由基。术语“卤化物”意指氟化物(F-)、氯化物(Cl-)、溴化物(Br-)或碘化物(I-)阴离子。除非本文另有指示,否则术语“杂原子”意指O、S、S(O)、S(O)2、Si(RC)2、Ge(RC)2、P(RP)或N(RN),其中独立地每个RC为未取代(C1-C40)烃基,每个RP为未取代(C1-C40)烃基;并且每个RN为未取代(C1-C40)烃基或不存在(当N包含-N=时不存在)。优选地,在式(I)的金属配体络合物中,除了S(O)或S(O)2双自由基官能团中的O-S键之外,不存在O-O、S-S或O-S键。更优选地,在式(I)的金属配体络合物中,除了S(O)或S(O)2双自由基官能团中的O-S键之外,不存在O-O、N-N、P-P、N-P、S-S或O-S键。优选地,在式(I)的金属配体络合物中,除了S(O)或S(O)2双自由基官能团中的O-S键之外,不存在O-O、S-S或O-S键。更优选地,在式(I)的金属配体络合物中,除了S(O)或S(O)2双自由基官能团中的O-S键之外,不存在O-O、N-N、P-P、N-P、S-S或O-S键。术语“饱和”意指缺少碳-碳双键,碳-碳三键和(在含杂原子的基团中)碳-氮、碳-磷和碳-硅双键或三键。当饱和化学基团由一个或多个取代基RS取代时,任选地取代基RS中可存在或可不存在一个或多个双键和/或三键。术语“不饱和”意指含有一个或多个碳-碳双键,碳-碳三键和(在含杂原子的基团中)碳-氮、碳-磷和碳-硅双键或三键,不包括任何可存在于取代基RS(如果有的话)或(杂)芳香环(如果有的话)中的此类双键。M是钛、锆或铪。在一个实施例中,M是锆或铪,并且在另一个实施例中,M是铪。在一些实施例中,M处于+2、+3或+4形式氧化态。在一些实施例中,n为0、1、2或3。每个X独立地为中性、单阴离子或双阴离子单齿配体;或两个X连在一起形成中性、单阴离子或双阴离子双齿配位体。以使式(I)的金属配体络合物总体上是中性的方式选择X和n。在一些实施例中,每个X独立地为单齿配位体。在一个实施例中,当有两个或更多个X单齿配体时,每个X是相同的。在一些实施例中,单齿配体是单阴离子配体。单阴离子配体的净形式氧化态为-1。每个单阴离子配体可独立地为氢化物、(C1-C40)烃基碳阴离子、(C1-C40)杂烃基碳阴离子、卤化物、硝酸根、碳酸根、磷酸根、硫酸根、HC(O)O-、(C1-C40)烃基C(O)O-、HC(O)N(H)-、(C1-C40)烃基C(O)N(H)-、(C1-C40)烃基C(O)N((C1-C20)烃基)-、RKRLB-、RKRLN-、RKO-、RKS-、RKRLp-或RMRKRLSi-,其中每个RK、RL和RM独立地为氢、(C1-C40)烃基或(C1-C40)杂烃基,或RK和RL连在一起形成(C2-C40)亚烃基或(C1-C40)杂亚烃基并且RM如上文所定义。助催化剂(Co-catalyst)组分在一些实施例中,包含式(I)的金属配体络合物的前催化剂可通过使其与活化助催化剂接触或组合或通过使用如本领域已知的供金属基烯烃聚合反应使用的活化技术而具有催化活性。适用于本文的活化助催化剂包括烷基铝;聚合物或低聚物铝氧烷(也称作铝氧烷);中性路易斯酸(Lewisacids);和非聚合物非配位离子形成型化合物(包括在氧化条件下使用此类化合物)。适合的活化技术是整体电解法(bulkelectrolysis)。还考虑了一种或多种前述活化助催化剂和技术的组合。术语“烷基铝”意指单烷基铝二氢化物或单烷基铝二卤化物、二烷基铝氢化物或二烷基铝卤化物、或三烷基铝。铝氧烷和其制备在例如美国专利号(UnitedStatesPatentNumber,USPN)US6,103,657中是已知的。优选的聚合物或低聚物铝氧烷的实例是甲基铝氧烷,三异丁基铝改性甲基铝氧烷和异丁基铝氧烷。示例性的路易斯酸活化助催化剂是含有1至3个如本文所述的烃基取代基的第13族金属化合物。在一些实施例中,示例性的第13族金属化合物是三(烃基)-取代铝或三(烃基)-硼化合物。在一些其它实施例中,示例性的第13族金属化合物是三(烃基)-取代铝或三(烃基)-硼化合物,是三((C1-C10)烷基)铝或三((C6-C18)芳基)硼化合物和其卤化(包括全卤化)衍生物。在一些其它实施例中,示例性的第13族金属化合物是三(氟取代的苯基)硼烷,在其它实施例中,是三(五氟苯基)硼烷。在一些实施例中,活化助催化剂是三((C1-C20)烃基)硼酸盐(例如,三苯甲基四氟硼酸盐)或三((C1-C20)烃基)铵四((C1-C20)烃基)硼烷(例如,双(十八烷基)甲基铵四(五氟苯基)硼烷)。如本文所用,术语“铵”意指氮阳离子,其为((C1-C20)烃基)4N+、((C1-C20)烃基)3N(H)+、((C1-C20)烃基)2N(H)2+、(C1-C20)烃基N(H)3+或N(H)4+,其中每个(C1-C20)烃基可相同或不同。中性路易斯酸活化助催化剂的示例性组合包括混合物,所述混合物包含三((C1-C4)烷基)铝和卤化三((C6-C18)芳基)硼化合物,特别是三(五氟苯基)硼烷的组合。其它示例性实施例是此类中性路易斯酸混合物与聚合物或低聚物铝氧烷的组合,以及单一中性路易斯酸,特别是三(五氟苯基)硼烷与聚合物或低聚物铝氧烷的组合。示例性实施例((金属配体络合物)∶(三(五氟苯基硼烷)∶(铝氧烷)[例如,(第4族金属配体配合物)∶(三(五氟苯基硼烷)∶(铝氧烷)]的摩尔数之比为1∶1∶1至1∶10∶30,其它示例性实施例为1∶1∶1.5至1∶5∶10。先前已在以下美国专利中关于不同金属配体络合物教示了许多活化助催化剂和活化技术:US5,064,802;US5,153,157;US5,296,433;US5,321,106;US5,350,723;US5,425,872;US5,625,087;US5,721,185;US5,783,512;US5,883,204;US5,919,983;US6,696,379;和US7,163,907。适合的烃基氧化物的实例公开于US5,296,433中。适用于加成聚合催化剂的布朗斯特酸盐(Bronstedacidsalts)的实例公开于US5,064,802;US5,919,983;US5,783,512中。适用于加成聚合催化剂的作为活化助催化剂的阳离子氧化剂和非配位相容性阴离子的盐的实例公开于US5,321,106中。适用于加成聚合催化剂的作为活化助催化剂的碳正离子盐的实例公开于US5,350,723中。适用于加成聚合催化剂的作为活化助催化剂的甲硅烷鎓(silylium)盐的实例公开于US5,625,087中。适合的醇、硫醇、硅醇和肟与三(五氟苯基)硼烷的络合物的实例公开于US5,296,433中。这些催化剂中的一些也描述于US6,515,155B1的在第50栏第39行开始并且直到第56栏第55行的一部分中,所述专利的仅这部分以引用的方式并入本文。在一些实施例中,包含式(I)的金属配体络合物的前催化剂可通过与一种或多种助催化剂(如阳离子形成型助催化剂、强路易斯酸或其组合)组合而经活化以形成活化催化剂组合物。适用的助催化剂包括聚合物或低聚物铝氧烷,特别是甲基铝氧烷,以及惰性相容性非配位离子形成型化合物。示例性的适合的助催化剂包括但不限于改性甲基铝氧烷(modifiedmethylaluminoxane,MMAO)、双(氢化牛脂烷基)甲基、四(五氟苯基)硼酸盐(1-)胺、三乙基铝(triethylaluminum,TEA)和其任何组合。在一些实施例中,前述活化助催化剂中的一种或多种以彼此组合的方式使用。特别优选的组合是三((C1-C4)烃基)铝、三((C1-C4)烃基)硼烷或硼酸铵与低聚物或聚合物铝氧烷化合物的混合物。一种或多种式(I)的金属配体络合物的总摩尔数与一种或多种活化助催化剂的总摩尔数之比为1∶10,000至100∶1。在一些实施例中,所述比为至少1∶5000,在一些其它实施例中,至少1∶1000;和10∶1或更小,并且在一些其它实施例中,1∶1或更小。当铝氧烷单独用作活化共催化剂时,优选地采用的铝氧烷的摩尔数为式(I)的金属配体络合物的摩尔数的至少100倍。当单独使用三(五氟苯基)硼烷作为活化助催化剂时,在一些其它实施例中,所采用的三(五氟苯基)硼烷的摩尔数比式(I)的一种或多种金属配体络合物的总摩尔数为0.5∶1至10∶1,在一些其它实施例中,形成,1∶1至6∶1,在一些其它实施例中,1∶1至5∶1。剩余的活化助催化剂通常以大约等于一种或多种式(I)的金属配体络合物的总摩尔量的摩尔量使用。最终用途应用根据本公开的LLDPE适用于流延膜挤出工艺或吹塑膜挤出工艺,其中膜至少在机器方向上进一步定向。根据本公开的LLDPE可以纯净形式或以与其它聚合物、添加剂和填充剂的共混物形式挤出。膜可为通过单个或多个模头的各种挤出获得的单层或共挤出多层膜。所得膜可以原样使用或可层压到其它膜或衬底上,例如通过热粘合剂层压或直接挤出到衬底上。所得膜和层压制件可经受其它成型操作,例如压花、拉伸、热成型。可施加如电晕的表面处理并且可印刷膜。根据本公开的由LLDPE制备的膜表现出陡峭的拉伸机器方向(machinedirection,MD)曲线,这反过来导致在机器方向定向(machinedirectionorientation,MDO)活化期间的优异拉伸性。根据本公开的由LLDPE制备的膜在机器方向定向期间的拉伸比可以为至少1.5X,例如,2X至6X,或替代地2X至5.5X,或替代地2X至5X,或替代从2X到4.5X。根据本发明的透气膜的基重在5至25gsm的范围内。本文包括并公开5至25gsm的所有单个值和子范围;例如膜的基重可在5、8、11、14、17、20或23gsm的下限至6、9、12、15、18、21或25gsm的上限范围内。例如,膜的基重可以在5至25gsm,或替代地,5至15,或替代地,15至25gsm,或替代地,10至20gsm,或替代地,从10至23gsm。根据本发明的透气膜的水蒸气透过率在1,000至9.000克每平方米每天(克/平方米-天)的范围内,在38℃下测量。本文包括并公开1,000至9,000克/平方米-天的所有单个值和子范围;例如,水蒸气透过率可在1,000、3,000或5,000克/平方米-天的下限到2,000、5000、8,000或9,000克/平方米-天的上限范围内。例如,水蒸气透过率可以在1,000至9,000克/平方米-天,或替代地,1,000至5,000克/平方米-天,或替代地,5,000至9,000克/平方米-天,或替代地,3,000至7,000克/平方米-天的范围内。由发明组合物制备的膜和层压制件可用于各种目的,例如食品包装。所述膜还适用于卫生和医疗应用,例如用于尿布、成人失禁产品、女性卫生产品、床衬垫、动物训练产品和动物失禁产品的透气膜。实例以下实例说明本发明但并不旨在限制本发明的范围。本发明的实例证明,选择本公开的LLDPE导致在机器方向定向期间的优越性能,即非常低的“最小拉伸比”,同时保持可接受的最终膜性质,如水蒸气透过率(watervaportransmissionrate,WVTR)、收缩率和即使在低拉伸比下的穿刺强度。比较组合物1是DOWLEX2107G是非均匀支化乙烯-辛烯共聚物,其熔融指数(I2)为约2.3克/10分钟、70至90℃的CEF级分为48.6%、I10/I2为8.5并且密度为0.917g/cm3,可购自陶氏化学公司(TheDowChemicalCompany)。比较组合物2是EXCEED是超过3518是在茂金属催化剂体系存在下经由气相聚合工艺制备的乙烯-己烯共聚物,其熔融指数(I2)为3.5克/10分钟、70至90℃的CEF级分为74.5%、I10/I2为5.8并且密度为0.918g/cm3,可购自ExxonMobilChemicalCompany。发明组合物1是乙烯-己烯共聚物,其熔融指数(I2)为3.2克/10分钟、70至90℃的CEF级分为91.9%、I10/I2为6.5并且密度为0.918g/cm3。发明组合物1在US专利美国专利5977251中描述的单环路反应器系统中在存在包含由下式表示的前催化剂的催化剂体系下经由溶液聚合制备:发明组合物1的聚合条件在表1和2中报告。参考表1和2,TEA是三乙基铝,并且PETROSOLD100/120是溶剂,其可商购自(dePetróleos,S.A.U.,Madrid,Spain)。测量发明组合物1和比较组合物1和2的性质并在表3-6中报告。表1表2发明组合物13.催化剂反应器助催化剂-1/催化剂摩尔进料比3.0反应器助催化剂-1型双(氢化牛脂烷基)甲基、四(五氟苯基)硼酸盐(1-)胺反应器助催化剂-2/催化剂摩尔比33反应器助催化剂-2型TEA表3表4Mw(g/mol)ZSV(Pas)ZSVR发明组合物17500028201.98比较组合物27710020501.30比较组合物18710041901.70表5Mn(g/mol)Mw(g/mol)Mz(g/mol)Mw/MnMz/Mw发明组合物134300750001360002.191.81比较组合物231000771001370002.491.77比较组合物121600871003270004.033.75表6结晶热(g/mol)峰值结晶温度(℃)熔化热(g/mol)峰值熔化温度(℃)发明组合物1139.797.1139.7112.3比较组合物2145.6101.1145.1113.8比较组合物1140.7107.1140.3123.3根据表7中所示的工艺条件,在Collin流延生产线上挤出发明组合物1和比较组合物1和2以形成单层膜。所述膜在本文中以形成膜的组合物的方式提及。表7模隙(mm):0.8线速度[米/分钟]:7.5熔融温度(℃):215产出率(Kg/h):5厚度(μm):50测量基于发明组合物1和比较组合物1和2的膜的性质并在图1和2中报告。参考图1,拉伸曲线清楚地指示由发明组合物1制备的膜比由比较组合物1和2制备的膜更快地到达应变硬化区。本文将200mm伸长率下的张力与75mm伸长率下的张力之比定义为硬化率。由发明组合物1制备的膜和由比较组合物1和2制备的膜的硬化比在表8中给出。表8-硬化率比较组合物1=1.08比较组合物2=1.08发明组合物1=1.22经由Buss配混机进一步配混发明组合物1和比较组合物1和2以包括50重量%的CaCO3。将得到的化合物,发明复合组合物1、比较复合组合物1和比较复合组合物2中的每一个分别在60℃下干燥六小时,并且然后装入铝袋中以避免在挤出前吸收水分。发明复合组合物1、比较复合组合物1和比较复合组合物2经由配备有机器方向定向(MDO)拉伸单元的Collin流延挤出生产线挤出,其中目标基重为18GSM。将化合物进料到料斗中并通过流延模头挤出成薄膜。然后通过以不同速度运行的加热辊在MDO单元中拉伸膜。所述装置允许拉伸膜和随后的膜退火。拉伸计算为最终速度(卷绕机)与MDO单元的引入辊速度之间的比率。在挤出和拉伸期间的工艺设置如下表9所示:表9熔融温度:225℃(进料区45℃、第一区190℃、第二区210℃、所有其它区:230℃)模隙:0.75mm42微米过滤器挤出机RPM35冷却辊处于45℃在60℃下拉伸,在40℃下引入和退火辊对于比较复合组合物1,增加拉伸比直至虎纹消失并且膜具有视觉均匀的外观。虎纹消失时的拉伸比称为“比较复合组合物1最小拉伸比”。在所述拉伸比下,通过调节MDO单元的引入辊处的速度获得18GSM的目标基重,并取出膜样品。然后运行比较复合组合物2。“比较复合组合物2最小拉伸比”类似于“比较复合组合物1最小拉伸比”。取出膜样品。接下来运行发明复合组合物1。“发明复合组合物1最小拉伸比”显著低于“比较复合组合物2最小拉伸比”。取出膜样品。然后增加拉伸比直至达到“比较复合组合物2最小拉伸比”的值,并取出第二膜样品。拉伸比在表10中报告。表10测量如上获得的表4中所示的四个膜样品的性质如图3-6所示:测量如表4中所示的四个膜样品中的每一个的断裂拉伸伸长率CD,并且结果在表11中给出。表11断裂拉伸伸长率CD比较复合组合物1/SR5503%比较复合组合物2/SR5.2496%发明复合组合物1/SR5.2481%发明复合组合物1/SR2.8499%参考图3,所述值之间的差异对应于可以用发明复合组合物1在2.8x至5.2x的拉伸范围内获得的性质窗口。在相同的拉伸比下,发明配混组合物1和对比配混组合物1和2落入相同的抗穿刺性范围内。参考图4,可以用发明化合物获得宽范围的水头值。比较相同拉伸比下发明复合组合物1与比较复合组合物1和2显示了发明复合组合物1的优越性。参考图5,在测试的拉伸比内,发明复合组合物1的WVTR窗口相对较窄,即在低拉伸比下也可以获得可接受的WVTR值。在相同拉伸比下,发明复合组合物1的WVTR略高比较复合组合物1的WVTR,并且与比较复合组合物2在相同范围内(差异<5%)。参考图6,在相同水平下拉伸的配置物之间的比较显示发明复合组合物1超越比较复合组合物1和2的更好的性能。出人意料地,参考图7,发明复合组合物1的收缩率似乎与拉伸比无关。在相同拉伸比下,发明复合组合物1的收缩率优于比较复合组合物1并且相对接近比较复合组合物2。测试方法测试方法包括以下各项:熔融指数根据ASTMD-1238在190℃下并且分别在2.16kg和10kg负载下测量熔融指数(I2和I10)。其值以克/10分钟为单位报告。密度根据ASTMD4703制备用于密度测量的样品。在使用ASTMD792、方法B压制样品一小时内进行测量。高温凝胶渗透色谱法凝胶渗透色谱(GelPermeationChromatography,GPC)系统由配备有板载差示折光仪(RI)(其它适合的浓度检测器可以包括来自PolymerChAR(西班牙瓦伦西亚(Valencia,Spain))的IR4红外检测器)的沃特世(Waters)(麻省米尔福德(Milford,Mass))150C高温色谱仪(其它适合的高温GPC仪器包括聚合物实验室(PolymerLaboratories)(英国什罗普郡(Shropshire,UK))型号210和型号220)组成。使用ViscotekTriSEC软件版本3和4通道Viscotek数据管理器DM400执行数据收集。所述系统还配备有来自聚合物实验室(英国什罗普郡)的在线溶剂脱气装置。可以使用适合的高温GPC柱,如四个30em长ShodexHT80313微米柱或20微米混合孔径填充物(MixALS,聚合物实验室)的四个30cm聚合物实验室柱。在140℃下操作样品转盘室,并且在150℃下操作柱室。以50毫升溶剂中0.1克聚合物的浓度制备样品。色谱溶剂和样品制备溶剂含有200ppm三氯苯(trichlorobenzene,TCB)。两种溶剂都用氮气喷射。在160℃下温和搅拌聚乙烯样品四小时。注射体积为200微升。将通过GPC的流率设定为1毫升/分钟。通过运行21个窄分子量分布聚苯乙烯(polystyrene,PS)标样来校准GPC柱组。标样的分子量(molecularweight,MW)在580至8,400,000的范围内,并且标样包含于6种“混合液(cocktail)”混合物中。每种标样混合物单个分子量之间的间隔为至少十倍。标准混合物购自聚合物实验室。对于分子量等于或大于1,000,000,以50mL溶剂中0.025g,并且对于分子量小于1,000,000,以50mL溶剂中0.05g制备聚苯乙烯标样。在80℃下温和搅拌30分钟使聚苯乙烯标样溶解。首先运行窄标样混合物并且按最高分子量组分降低的次序以使降级最小化。使用以下等式将聚苯乙烯标样峰值分子量转化成聚乙烯分子量(如在Williams和Ward,《聚合物科学杂志(J.Polym.Sci.)》,《聚合物通讯(Polym.Letters)》,6,621(1968年)中所述):M聚乙烯=A×(M聚苯乙烯)B,其中M为聚乙烯或聚苯乙烯的分子量(如所标记的),并且B等于1.0。本领域普通技术人员已知A可在约0.38至约0.44的范围内,并且在使用宽聚乙烯标样校准时测定。使用这种聚乙烯校准方法来获得分子量值,如分子量分布(molecularweightdistribution,MWD或Mw/Mn),和相关统计(通常是指常规GPC或cc-GPC结果)在此定义为改进的Williams和Ward方法。DSC结晶度差示扫描量热法(DifferentialScanningcalorimetry,DSC)可以用于测量聚合物在广泛温度范围内的熔融和结晶行为。例如,使用配备有RCS(refrigeratedcoolingsystem,冷藏冷却系统)和自动取样器的TA仪器(TAInstruments)Q1000DSC来执行这一分析。在测试期间,使用50毫升/分钟的氮气吹扫气流。在约175摄氏度下将每个样品熔融压制成薄膜;然后将熔融样品空气冷却至室温(约25℃)。从冷却的聚合物中抽取3-10mg、6mm直径试样,称重、放置于轻铝盘(约50mg)中,并关闭。然后执行分析以测定其热特性。通过使样品温度缓慢上升和下降以产生热流对温度廓线来测定样品的热行为。首先,将样品快速加热到180℃,并且保持等温达3分钟以去除其热过程(thermalhistory)。接着,以10℃/分钟的冷却速率将样品冷却到-40℃,并且在-40℃下保持等温达3分钟。然后以10℃/分钟的加热速率将样品加热到150℃(这是“第二加热”缓变)。记录冷却和第二加热曲线。通过将基线端点(baselineendpoint)设定为从开始结晶至-20℃来分析冷却曲线。通过将基线端点设定为从-20℃到熔融结束来分析加热曲线。测定的值为峰值熔融温度(Tm)、峰值再结晶温度(Tp)、熔解热(heatoffusion)(Hf)(以焦耳/克为单位)和使用以下等式计算的聚乙烯样品的%结晶度:%结晶度=((Hf)/(292J/g))×100。熔解热(Hf)和峰值熔融温度从第二加热曲线中报告。由冷却曲线确定峰值再结晶温度为Tp。结晶洗脱分级(CrystallizationElutionFractionation,CEF)方法结晶洗脱分级(CEF)方法根据Monrabal等人,大分子研讨会(Macromol.Symp.)》257,71-79(2007)中描述的方法进行,其以引用的方式并入本文。CEF仪器装备有IR-4检测器(如西班牙的PolymerChar商业上出售的那些)和双角度光散射检测器型号2040(如PrecisionDetectors商业上出售的那些)。IR-4检测器在组合模式下用两个过滤器操作:C006和B057。50mm×4.6mm的10微米保护柱(如聚合物实验室商业上出售的那些)安装在检测器烘箱中的IR-4检测器前。获得邻二氯苯(Ortho-dichlorobenzene,ODCB,99%无水级)和2,5-二-叔丁基-4-甲基苯酚(BHT)(如可商购自西格玛奥德里奇(Sigma-Aldrich))。还获得硅胶40(粒度0.2~0.5mm)(如可商购自EMDChemicals)。使用前将硅胶在真空烘箱中在160℃下干燥约两小时。将八百毫克BHT和五克硅胶添加到两升ODCB中。含有BHT和硅胶的ODCB在下文中称作“ODCB-m”。使用前用干燥氮气(N2)鼓泡ODBC-m一小时。在<90psig下传送氮气越过CaCO3和分子筛来获得干燥氮气。通过使用自动取样器,将聚合物样品在160℃下在振动下以4mg/ml的速率溶解于ODCB-m中达2小时来制备样品溶液。将300μL样品溶液注射到柱中。CEF的温度廓线为:从110℃至25℃以3摄氏度/分钟的速率结晶、热平衡在30℃下达5分钟(包括可溶性级分洗脱时间设定为2分钟),和从25℃至140℃以3摄氏度/分钟的速率洗脱。结晶期间的流率为0.052毫升/分钟。洗脱期间的流率为0.50毫升/分钟。以一个数据点/秒收集IR-4信号数据。CEF柱在125μm±6%处填充有玻璃珠粒(如可商购自MO-SCI特殊产品(SpecialtyProducts)的那些),具有根据美国2011/0015346A1的1/8英寸不锈钢管。CEF柱的内部液体体积在2.1ml与2.3ml之间。通过使用NIST标准参考材料线性聚乙烯1475a(1.0mg/ml)和二十烷(2mg/ml)的混合物在ODCB-m中执行温度校准。校准由以下四个步骤组成:(1)计算定义为测量的二十烷峰值洗脱温度减去30.00℃之间的温度偏移的延迟体积(delayvolume);(2)从CEF原始温度数据中减去洗脱温度的温度偏移。应注意,所述温度偏移是实验条件,如洗脱温度、洗脱流率等的函数;(3)产生历经25.00℃和140.00℃范围的转换洗脱温度的线性校准线,使得NIST线性聚乙烯1475a的峰值温度为101.00℃,并且二十烷的峰值温度为30.00℃,(4)对于在30℃下等温测量的可溶性级分,通过使用3摄氏度/分钟的洗脱加热速率来线性外推洗脱温度。获得报告的洗脱峰值温度,使得观测的共聚单体含量校准曲线与美国8,372,931中先前报告的那些一致。根据以下等式,70至90℃的CEF级分定义为IR-4色谱图(基线减去测量通道)在70.0至90.0℃范围内的洗脱温度的积分除以25至140.0℃的总积分:其中T为洗脱温度(来自上面论述的校准)。通过选择两个数据点计算线性基线:一个在聚合物洗脱之前,通常在25.5℃温度下,并且另一个在聚合物洗脱之后,通常在118℃下。对于每个数据点,在积分之前从基线减去检测器信号。蠕变零剪切粘度测量方法零剪切粘度经由蠕变测试获得,其在AR-G2应力控制流变仪(TA仪器;特拉华州纽卡斯尔(NewCastle,Del))上使用25mm直径平行板在190℃下进行。在将固定装置归零之前,将流变仪烘箱设定为测试温度达至少30分钟。在测试温度下,将压缩模制的样品盘插入板之间并允许达到平衡5分钟。然后将上板向下降低到所需测试间隙(1.5mm)以上50μm。修剪掉任何多余材料,并将上板降低到所需间隙。在氮气以5升/分钟的流率吹扫下进行测量。将默认蠕变时间设定为2小时。对所有样品施加20Pa恒定低剪切应力,以确保稳态剪切速率足够低以处于牛顿区(Newtonianregion)中。对于本研究中的样品,所得稳态剪切速率在10-3至10-4s-1的范围内。通过对log(J(t))对log(t)曲线的最后10%时间窗口中的所有数据进行线性回归确定稳态,其中J(t)为蠕变柔量,并且t为蠕变时间。如果线性回归的斜率大于0.97,则认为达到稳态,然后停止蠕变测试。在本研究中的所有情况下,斜率在2小时内符合标准。由ε对t曲线的最后10%时间窗口中所有数据点的线性回归的斜率确定稳态剪切速率,其中ε为应变。由施加的应力与稳态剪切速率之比确定零剪切粘度。为了确定样品在蠕变测试期间是否降级,从0.1至100rad/s在相同试样上在蠕变测试之前和之后进行小振幅振荡剪切测试。比较两个测试的复数粘度值。如果0.1rad/s下的粘度值差异大于5%,则认为样品在蠕变测试期间已经降级,并且废弃结果。零剪切粘度比(ZSVR)定义为根据以下等式,在当量重均分子量(Mw-gpc)下,支化聚乙烯材料的零剪切粘度(zero-shearviscosity,ZSV)与线性聚乙烯材料的ZSV之比:ZSV值经由上述方法从190℃下的蠕变测试中获得。Mw-gpc值通过常规GPC方法确定。基于一系列线性聚乙烯参比材料建立线性聚乙烯的ZSV与其Mw-gpc之间的相关性。ZSV-Mw关系的描述可以在ANTEC程序中找到:Kariala、TeresaP.;Sammler、RobertL.;Mangnus、MarcA.;Hazlitt、LonnieG.;Johnson、MarkS.;Hagen、CharlesM.、Jr.;Huang、JoeW.L.;Reichek、KennethN.检测聚烯烃中的低水平长链支化。年度技术会议-塑料工程师协会(AnnualTechnicalConference-SocietyofPlasticsEngineers)(2008),第66届887-891。1HNMR方法在10mmNMR管中,将3.26g储备溶液加入0.133g聚烯烃样品中。储备溶液是四氯乙烷-d2(tetrachloroethane,TCE)和全氯乙烯(50∶50,w∶w)与0.001MCr3+的混合物。将管中的溶液用N2吹扫5分钟以减少氧气量。将封盖的样品管留在室温下过夜以溶胀聚合物样品。将样品在110℃下振动溶解。样品不含可能促成不饱和度的添加剂,例如增滑剂,如芥酸酰胺。1HNMR在BrukerAVANCE400MHz光谱仪上在120℃下用10mm冷冻探针运行。运行两个实验以获取不饱和度:对照和双预饱和实验。对于对照实验,用LB=1Hz的指数窗函数处理数据,从7至-2ppm校正基线。将TCE的残余物的1H信号设定为100,在-0.5至3ppm的积分Itotal用作对照实验中整个聚合物的信号。如下计算聚合物中的CH2基团数NCH2:NCH2=Itotal/2对于双预饱和实验,用LB=1Hz的指数窗函数处理数据,从6.6至4.5ppm校正基线。将TCE的残余物的1H信号设定为100,基于下图所示的区域对不饱和度的相应积分(I次亚乙烯基、I三取代、I乙烯基和I亚乙烯基)进行积分计算次亚乙烯基、三取代、乙烯基和亚乙烯基的不饱和单元数:N次亚乙烯基=I次亚乙烯基/2N三取代=I三取代N乙烯基=I乙烯基/2N亚乙烯基=I亚乙烯基/2如下计算不饱和单元/1,000,000碳:N次亚乙烯基/1,000,000C=(N次亚乙烯基/NCH2)*1,000,000N三取代/1,000,000C=(N三取代/NCH2)*1,000,000N乙烯基/1,000,000C=(N乙烯基/NCH2)*1,000,000N亚乙烯基/1,000,000C=(N亚乙烯基/NCH2)*1,000,000不饱和度NMR分析的要求包括:Vd2的定量水平为0.47±0.02/1,000,000碳,用200次扫描(数据采集小于1小时,包括运行对照实验的时间)其中样品为3.9重量%(对于Vd2结构而言,见《大分子(Macromolecules)》,2005年,第38期,6988),10mm高温冷冻探针。定量水平定义为10信噪比。将TCT-d2的残余质子的1H信号的化学位移参考设定为6.0ppm。对照使用ZG脉冲、TD32768、NS4、DS12、SWH10,000Hz、AQ1.64s、D114s运行。双预饱和实验用修改的脉冲序列运行,O1P1.354ppm、O2P0.960ppm、PL957db、PL2170db、TD32768、NS200、DS4、SWH10,000Hz,AQ1.64s、D11s、D1313s。使用BrukerAVANCE400MHz光谱仪的用于不饱和的修改的脉冲序列如下所示:膜测试条件在所制备的膜上测量以下物理特性:拉伸测试:ISO527-3收缩率ASTMD2732穿刺:ASTMD-5748水头:ISO1420水蒸气透过率(WVTR):ASTME398(在Lissy上测量)可以在不脱离本发明要旨和基本属性的情况下以其它形式实施本发明,并且因此,应参考所附权利要求书,而非前述说明书来指示本发明的范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1