具有JAK抑制作用的化合物的晶体的制作方法
本发明涉及具有JAK抑制作用的新型化合物。
背景技术:
酪氨酸激酶是将蛋白质的酪氨酸残基特异性地磷酸化的酶,在细胞内信号传导系统中起着重要的作用,参与细胞的生存、分化、增殖、分泌等广泛的生物学机能。作为与细胞因子信号传导相关的细胞内酪氨酸激酶,已知两面神激酶(Janus Kinase,也称为JAK)家族。JAK家族中存在JAK1、JAK2、JAK3、酪氨酸激酶2(Tyrosine Kinase 2,也称为Tyk2)4种酶。通过细胞因子与细胞因子受体结合,JAK被磷酸化,将受体的酪氨酸残基磷酸化。然后,细胞内存在的信号传导及转录激活因子(signal transducer and activator of transcription,也称为STAT)与被磷酸化的受体的酪氨酸残基缔合,STAT的酪氨酸残基通过JAK被磷酸化。认为被磷酸化的STAT形成二聚体,转移至核内,作为转录因子诱导目标基因的转录活化,引起细胞的活化。JAK/STAT系统是免疫活性细胞中的细胞因子的最主要的细胞内信号传导系统(非专利文献1),通过4种JAK与7种STAT的组合,能进行约40种细胞因子的信号传导,因此认为,细胞因子产生的异常和细胞因子信号传导的异常不仅与自身免疫性疾病、过敏等各种免疫疾病和炎症疾病有关,而且与癌症等具有多种不同病理的疾病有很大关联。这些抑制JAK/STAT系统活化的化学物质作为这些疾病的新的治疗药受到关注,事实上,JAK抑制剂作为骨髓纤维化、真性红细胞增多症和类风湿性关节炎的治疗药已经在美国和日本等被认可。此外,也期待对其它自身免疫性疾病(例如牛皮癣关节炎、幼年型关节炎、卡斯特曼病、全身性红斑狼疮、斯耶格伦综合征、多发性硬化症、炎性肠病、贝切特病、重症肌无力、I型糖尿病、免疫球蛋白肾病、自身免疫性甲状腺病、银屑病、硬皮病、狼疮性肾炎、干眼症、血管炎(例如大动脉炎、巨细胞动脉炎、显微镜下多血管炎、肉芽肿性多血管炎、嗜酸性肉芽肿性多血管炎)、皮肤肌炎、多肌炎、视神经脊髓炎)、炎症性疾病(例如特应性皮炎、接触性皮炎、湿疹、瘙痒症、食物过敏、支气管哮喘、嗜酸性粒细胞性肺炎、慢性阻塞性肺病、过敏性鼻炎、慢性鼻窦炎、嗜酸性粒细胞性鼻窦炎、鼻息肉、过敏性结膜炎、骨关节炎、强直性脊柱炎、川崎病、伯格氏病、结节性多动脉炎、IgA血管炎)、增殖性疾病(例如实体癌、血液癌、淋巴系统恶性肿瘤、骨髓增殖性疾病、多发性骨髓瘤、肺纤维化、嗜酸性粒细胞增多症)、突发性聋、糖尿病肾病、斑秃、骨髓移植排异或脏器移植排异等的治疗效果,现在,上述疾病中的一些疾病首先在日本、并且也在美国和欧洲实施了临床试验。
各种生物学研究表明,其中的JAK1具有与多种细胞因子的信号传导相关的重要作用(参照非专利文献2、3、4),这就表示JAK1抑制剂对于例如自身免疫性疾病:牛皮癣关节炎(例如参照非专利文献5)、幼年型关节炎(例如参照非专利文献6)、卡斯特曼病(例如参照非专利文献6)、全身性红斑狼疮(例如参照非专利文献7)、斯耶格伦综合征(例如参照非专利文献8)、多发性硬化症(例如参照非专利文献9)、炎性肠病(例如参照非专利文献10)、贝切特病(例如参照非专利文献11)、重症肌无力(例如参照非专利文献12)、I型糖尿病(例如参照非专利文献9)、免疫球蛋白肾病(例如参照非专利文献13)、自身免疫性甲状腺病(例如参照非专利文献14)、银屑病(例如参照非专利文献15)、硬皮病(例如参照非专利文献16)、狼疮性肾炎(例如参照非专利文献17)、干眼症(例如参照非专利文献18)、血管炎(例如参照非专利文献19、20、21、22、23)、皮肤肌炎(例如参照非专利文献24)、多肌炎(例如参照非专利文献24)、视神经脊髓炎(例如参照非专利文献25)、炎症性疾病:特应性皮炎(例如参照非专利文献26)、接触性皮炎(例如参照非专利文献27)、湿疹(例如参照非专利文献28)、瘙痒症(例如参照非专利文献29)、食物过敏(例如参照非专利文献30)、支气管哮喘(例如参照非专利文献31)、嗜酸性粒细胞性肺炎(例如参照非专利文献32)、慢性阻塞性肺病(例如参照非专利文献33)、过敏性鼻炎(例如参照非专利文献31)、慢性鼻窦炎(例如参照非专利文献34)、嗜酸性粒细胞性鼻窦炎、鼻息肉(例如参照非专利文献35)、过敏性结膜炎(例如参照非专利文献36)、骨关节炎(例如参照非专利文献37)、强直性脊柱炎(例如参照非专利文献6)、川崎病(例如参照非专利文献38)、伯格氏病(例如参照非专利文献39)、结节性多动脉炎(例如参照非专利文献40)、IgA血管炎(例如参照非专利文献41)等、增殖性疾病:实体癌、血液癌、淋巴系统恶性肿瘤、骨髓增殖性疾病、多发性骨髓瘤(例如参照非专利文献42、43、44)、突发性聋(例如参照非专利文献45)、糖尿病肾病(例如参照非专利文献46)、斑秃(例如参照非专利文献47)、骨髓移植排异或脏器移植排异等疾病的治疗有用,例如实施了以下的临床试验。
(1)类风湿性关节炎(https://clinicaltrials.gov/NCT01888874、NCT02049138)
(2)克隆氏病(https://clinicaltrials.gov/NCT02365649)
(3)非小细胞肺癌(https://clinicaltrials.gov/NCT02257619)
(4)胰腺癌(https://clinicaltrials.gov/NCT01858883)
(5)骨髓纤维化(https://clinicaltrials.gov/NCT01633372)
(6)银屑病(https://clinicaltrials.gov/NCT02201524)
此外,JAK1相关的细胞因子信号传导中,以下的细胞因子抑制剂已经上市。
(1)IL-6(也称为白细胞介素-6):类风湿性关节炎、幼年型关节炎和卡斯特曼病治疗药(参照非专利文献48、49、50)。
(2)IL-2:肾移植后的急性排异反应的治疗药(参照非专利文献51)。
现在还在实施以下细胞因子抑制剂临床试验。
(3)IL-4和IL-13:支气管哮喘、特应性皮炎、嗜酸性粒细胞性鼻窦炎、鼻息肉和嗜酸性粒细胞性食管炎治疗药(参照非专利文献31)。
(4)IL-13:肺纤维化治疗药(https://clinicaltrials.gov/NCT02036580)。
(5)IL-5:支气管哮喘、慢性阻塞性肺病、嗜酸性粒细胞增多症、嗜酸性肉芽肿性多血管炎、嗜酸性粒细胞性食管炎、嗜酸性粒细胞性鼻窦炎、鼻息肉和特应性皮炎治疗药(参照非专利文献31、非专利文献52)。
(6)IFNα(也称为干扰素-α):全身性红斑狼疮治疗药(参照非专利文献7)。
(7)IL-31:特应性皮炎治疗药(https://clinicaltrials.gov/NCT01986933)。
(8)TSLP(Thymic stromal lymphopoietin:也称为胸腺基质淋巴细胞生成素):支气管哮喘(https://clinicaltrials.gov/NCT02054130)、特应性皮炎治疗药(https://clinicaltrials.gov/NCT00757042)。
因此,JAK1信号传导的抑制是预防或治疗自身免疫性疾病、炎症性疾病和增殖性疾病等由JAK1信号传导异常引起的疾病的理想方式。
作为具有JAK抑制活性的化合物,报道了[1,2,4]三唑并[1,5-a]吡啶化合物(例如参照专利文献1、2)、3环性吡嗪酮化合物(例如参照专利文献3)、吡咯并嘧啶化合物(例如参照专利文献4~7)、酞嗪化合物(例如参照专利文献8)、咪唑并吡咯并吡啶化合物(例如参照专利文献9、非专利文献53)、二氨基-1,2,4-三唑化合物(例如参照非专利文献54)、吡唑并[1,5-a]吡啶(例如参照专利文献10)、咪唑并[1,2-a]吡啶(例如参照专利文献11、12)、苯并咪唑化合物(例如参照专利文献13)、7-氮杂吲哚化合物(例如参照专利文献14)等,但其中均未公开本发明化合物。
现有技术文献
非专利文献
非专利文献1:O’Shea等,Immunity,2012,36,542-550.
非专利文献2:O’Sullivan等,Mol.Immunol.,2007,44,2497-2506.
非专利文献3:Quintás-Cardama等,Nat.Rev.Drug Discov.,2011,10,127-140.
非专利文献4:Haan等,Chem.Biol.,2011,18,314-323.
非专利文献5:Gan等,BioDrugs,2013,27,359-373.
非专利文献6:Mihara等,Clin.Sci.(Lond.),2012,122,143-159.
非专利文献7:Wallace等,71st Ann.Meet.Am.Coll.Rheumatol.,2007,Abs.1315.
非专利文献8:Gliozzi等,J.Autoimmun.,2013,40,122-133.
非专利文献9:Neurath等,Cytokine Growth Factor Rev.,2011,22,83-89.
非专利文献10:Vuitton等,Curr.Drug Targets,2013,14,1385-1391.
非专利文献11:Akdeniz等,Ann.Acad.Med.Singapore,2004,33,596-599.
非专利文献12:Dalakas,Ann.N.Y.Acad.Sci.,2012,1274,1-8.
非专利文献13:Goto等,Clin.Immunol.,2008,126,260-269.
非专利文献14:Nanba等,Thyroid,2009,19,495-501.
非专利文献15:Strober等,Br.J.Dermatol.,2013,169,992-999.
非专利文献16:Christner等,Curr.Opin.Rheumatol.,2004,16,746-752.
非专利文献17:Dong等,Lupus,2007,16,101-109.
非专利文献18:Lim等,Cornea,2015,34,248-252.
非专利文献19:Saadoun等,Arthritis Rheumatol.,2015,67,1353-1360.
非专利文献20:Kieffer等,Rev.Med.Interne.,2014,35,56-59.
非专利文献21:Takenaka等,Clin.Rheumatol.,2014,33,287-289.
非专利文献22:Kobold等,Clin.Exp.Rheumatol.,1999,17,433-440.
非专利文献23:Vaglio等,Allergy,2013,68,261-273.
非专利文献24:Gono等,Rheumatology,2014,53,2196-2203.
非专利文献25:Araki等,Neurology,2014,82,1302-1306.
非专利文献26:Bao等,JAKSTAT,2013,2,e24137.
非专利文献27:Takanami-Ohnishi等,J.Biol.Chem.,2002,277,37896-37903.
非专利文献28:Antoniu,Curr.Opin.Investig.Drugs,2010,11,1286-1294.
非专利文献29:Sokolowska-Wojdylo等,J.Eur.Acad.Dermatol.Vener eol.,2013,27,662-664.
非专利文献30:Brown等,Eur.Food Res.Technol.,2012,235,971-980.
非专利文献31:Legrand等,J.Allergy Clin.Immunol.Pract.,2015,3,167-174.
非专利文献32:Kita等,Am.J.Respir.Crit.Care Med.,1996,153,1437-1441.
非专利文献33:Southworth等,Br.J.Pharmacol.,2012,166,2070-2083.
非专利文献34:Van Zele等,Allergy,2006,61,1280-1289.
非专利文献35:Nabavi等,Allergol.Immunopathol.(Madr.),2014,42,465-471.
非专利文献36:Sakai等,Curr.Eye Res.,2013,38,825-834.
非专利文献37:Beekhuizen等,Eur.Cell Mater.,2013,26,80-90.
非专利文献38:Abe,Nihon Rinsho,2014,72,1548-1553.
非专利文献39:Slavov等,Clin.Exp.Rheumatol.,2005,23,219-226.
非专利文献40:Kawakami等,Acta.Derm.Venereol.2012,92,322-323.
非专利文献41:Gülhan等,Pediatr.Nephrol.,2015,30,1269-1277.
非专利文献42:Costa-Pereira等,Am.J.Cancer Res.,2011,1,806-816.
非专利文献43:Vainchenker等,Semin.Cell Dev.Biol.,2008,19,385-393.
非专利文献44:Li等,Neoplasia,2010,12,28-38.
非专利文献45:Masuda等,Otol.Neurotol.,2012,33,1142-1150.
非专利文献46:Donate-Correa等,J.Diabetes Res.,2015,948417.
非专利文献47:Zhang等,Arch.Dermatol.Res.,2015,307,319-331.
非专利文献48:Nishimoto等,J.Rheumatol.,2003,30,1426-1435.
非专利文献49:Yokota等,LANCET,2008,371,998-1006.
非专利文献50:Nishimoto等,Blood,2000,95,56-61.
非专利文献51:Nashan等,LANCET,1997,350,1193-1198.
非专利文献52:Kouro等,Int.Immunol.,2009,21,1303-1309.
非专利文献53:Kulagowski等,Journal of Medicanal Chemistry,2012,55,5901-5921.
非专利文献54:Malerich等,Bioorg.Med.Chem.Lett.,2010,20,7454-7457.
专利文献
专利文献1:WO2010/149769号
专利文献2:WO2010/010190号
专利文献3:WO2012/085176号
专利文献4:WO2009/114512号
专利文献5:WO2011/075334号
专利文献6:WO2012/022045号
专利文献7:WO2012/054364号
专利文献8:WO2012/037132号
专利文献9:WO2011/086053号
专利文献10:WO2011/101161号
专利文献11:WO2011/076419号
专利文献12:JP2011/136925号
专利文献13:WO2005/066156号
专利文献14:WO2007/084557号
技术实现要素:
发明所要解决的技术问题
本发明的目的在于提供一种具有优异的JAK1抑制作用的化合物。
解决技术问题所采用的技术方案
本发明人经过认真研究,结果发现下述化合物(说明书中有时称为本发明化合物)具有优异的JAK1抑制作用,从而完成了本发明。
即,本发明可例举以下(I)~(III)的发明。
(I)以下(1)~(6)中任一项所述的化合物。
(1)[1-({6-[(2S)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯甲苯磺酸盐一水合物、
(2)[1-({6-[(2R)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯甲苯磺酸盐一水合物、
(3)(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸甲酯甲苯磺酸盐一水合物、
(4)(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸乙酯、
(5)N-(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)环丙烷羧酰胺、
(6)N-(1-{[6-{[(2R)-3,3-二甲基丁烷-2-基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)环丙烷羧酰胺。
(II)以下(1)~(6)中任一项所述的晶体。
(1)使用Cu Kα射线而得的粉末X射线衍射图中,至少在以下衍射角2θ:12.6度、13.3度、17.3度、20.0度、20.4度、21.3度及22.3度处显示出衍射峰的[1-({6-[(2S)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯甲苯磺酸盐一水合物的晶体、
(2)使用Cu Kα射线而得的粉末X射线衍射图中,至少在以下衍射角2θ:12.6度、13.3度、17.3度、20.0度、20.4度、21.3度及22.3度处显示出衍射峰的[1-({6-[(2R)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯甲苯磺酸盐一水合物的晶体、
(3)使用Cu Kα射线而得的粉末X射线衍射图中,至少在以下衍射角2θ:12.6度、13.3度、17.2度、20.6度及21.8度处显示出衍射峰的(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸甲酯甲苯磺酸盐一水合物的晶体、
(4)使用Cu Kα射线而得的粉末X射线衍射图中,至少在以下衍射角2θ:12.0度、13.8度、15.0度、16.0度、19.4度、20.9度及21.9度处显示出衍射峰的(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸乙酯的晶体、
(5)使用Cu Kα射线而得的粉末X射线衍射图中,至少在以下衍射角2θ:11.1度、12.9度、15.4度、17.8度、21.2度及22.3度处显示出衍射峰的N-(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)环丙烷羧酰胺的晶体、
(6)使用Cu Kα射线而得的粉末X射线衍射图中,至少在以下衍射角2θ:10.6度、13.0度、14.6度、17.4度、17.7度、21.3度及21.7度处显示出衍射峰的N-(1-{[6-{[(2R)-3,3-二甲基丁烷-2-基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)环丙烷羧酰胺的晶体。
(III)含有(I)或(II)中任一项所述的化合物作为有效成分的药物组合物。
确定本发明的实施例及权利要求书中的衍射峰的衍射角2θ时,所得的值应理解为在该值的±0.2度的范围内,较好是在该值的±0.1度的范围内。
发明的效果
本发明的晶体纯度高,操作也容易,可以用作工业上制造的药物、即JAK抑制剂的制造原料药。
附图说明
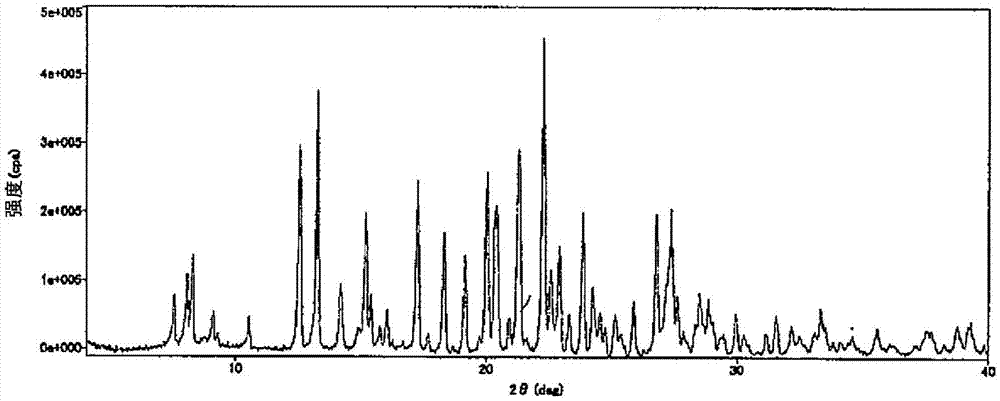
图1所示为[1-({6-[(2S)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯甲苯磺酸盐一水合物的晶体的粉末X射线衍射图(包含衍射角2θ:12.6度、13.3度、15.2度、17.3度、18.3度、19.1度、20.0度、20.4度、21.3度、22.3度、23.8度、26.8度、27.4度)。纵轴表示峰强度(cps),横轴表示衍射角(2θ[°])。
图2所示为[1-({6-[(2R)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯甲苯磺酸盐一水合物的晶体的粉末X射线衍射图(包含衍射角2θ:12.6度、13.3度、15.2度、17.3度、18.3度、19.2度、20.0度、20.4度、21.3度、22.3度、23.8度、26.8度、27.4度)。纵轴表示峰强度(cps),横轴表示衍射角(2θ[°])。
图3所示为(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸甲酯甲苯磺酸盐一水合物的晶体的粉末X射线衍射图(包含衍射角2θ:12.6度、13.3度、15.2度、17.2度、19.1度、20.1度、20.6度、21.8度、23.0度、24.0度、26.9度、27.2度)。纵轴表示峰强度(cps),横轴表示衍射角(2θ[°])。
图4所示为(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸乙酯的晶体的粉末X射线衍射图(包含衍射角2θ:12.0度、13.8度、15.0度、16.0度、17.7度、18.6度、19.4度、19.6度、20.2度、20.9度、21.9度、22.7度、24.1度)。纵轴表示峰强度(cps),横轴表示衍射角(2θ[°])。
图5所示为N-(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)环丙烷羧酰胺的晶体的粉末X射线衍射图(包含衍射角2θ:11.1度、11.5度、12.9度、15.4度、17.8度、18.3度、18.5度、21.2度、22.3度、24.3度、25.2度)。纵轴表示峰强度(cps),横轴表示衍射角(2θ[°])。
图6所示为N-(1-{[6-{[(2R)-3,3-二甲基丁烷-2-基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)环丙烷羧酰胺的晶体的粉末X射线衍射图(包含衍射角2θ:10.6度、13.0度、14.6度、17.4度、17.7度、20.8度、21.3度、21.7度、22.7度、25.0度、26.5度)。纵轴表示峰强度(cps),横轴表示衍射角(2θ[°])。
具体实施方式
以下,对本说明书中使用的各用语的定义进行详述。
作为“烷基”,可例举例如直链或支链的具有1个~10个碳原子、优选1个~8个碳原子、更优选1个~6个碳原子的烷基。具体可例举例如甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基、仲戊基、1-乙基丙基、1,2-二甲基丙基、叔戊基、2-甲基丁基、异戊基、新戊基、正己基、仲己基、1-乙基丁基、异己基、新己基、1,1-二甲基丁基、1,1,2-三甲基丙基、2-乙基丁基、1,2,2-三甲基丙基、2,2-二甲基丁基、庚基、异庚基、辛基、异辛基等。
作为“环烷基”,可例举例如碳数为3个~10个的1~3环性的饱和烃基。优选碳数为3个~6个的单环的环烷基。具体可例举环丙基、环丁基、环戊基、环己基、环庚基、二环[2.1.0]戊基、二环[2.2.1]庚基和二环[2.2.2]辛基等。
作为“芳基”,可例举例如1~3环性的、碳数为6个~14个的芳香族烃基。具体可例举苯基、1-萘基、2-萘基、1-蒽基、2-蒽基、9-蒽基、1-菲基、2-菲基、3-菲基、4-菲基、10-菲基等。其中,优选苯基。
本发明化合物可以按照例如以下所述的方法、后述的实施例或公知的方法,从公知化合物或能容易合成的中间体来制造。本发明化合物的制造中,原料具有对反应有影响的取代基的情况下,通常是预先通过公知的方法用适当的保护基将原料保护后进行反应。保护基在反应后可通过公知的方法除去。
制法1
[化1]
(上述反应式中,R1表示烷基或经烷基取代的环烷基,R2表示烷基或可以被烷基取代的芳基,R3表示烷基或被烷基取代的环烷基。)
工序1
本工序是将化合物1还原、从而得到化合物2的工序。
作为本反应中使用的还原剂,可使用硼氢化钠、三乙酰氧基硼氢化钠等。
还原剂优选以相对于化合物1为0.25~3倍摩尔当量的量使用。
作为本反应中使用的溶剂,只要不参与反应就没有特别限定,可例举例如四氢呋喃(以下称为“THF”)、乙醚等醚类,甲醇、乙醇等醇类,或者它们的混合溶剂。
反应温度为-78℃~100℃、优选为-30℃~20℃。
反应时间根据反应温度等而不同,通常为10分钟~24小时。
工序2
本工序是将化合物2用酸脱水后、与化合物3反应而环化、从而得到化合物4的工序。
作为本反应中使用的酸,可例举硫酸、甲磺酸、对甲苯磺酸等。
酸优选以相对于化合物2为1~3倍摩尔当量的量使用。
作为本反应中使用的溶剂,只要不参与反应就没有特别限定,可例举例如2-丙醇、乙醇等醇类,乙腈、丙腈等腈类,或者它们的混合溶剂。
反应温度为0℃~100℃、优选为20℃~70℃。
反应时间根据反应温度等而不同,脱水通常为10分钟~2小时,环化通常为30分钟~5小时。
工序3
本工序是将腈转化为亚氨酸酯的工序,是在碱金属醇盐之类的碱或氯化氢之类的酸的存在下在合适的溶剂中进行搅拌、从而得到化合物5的工序。
作为本反应中使用的碱,可例举甲醇钠、乙醇钠之类的醇盐类,作为酸,可例举氯化氢气体。此外,也可以由乙酰氯之类的酰氯和甲醇、乙醇之类的醇类来制备氯化氢。
碱和酸优选以相对于化合物4为1~100倍摩尔当量的量使用。
作为所使用的溶剂,只要不参与反应就没有特别限定,可例举例如甲醇、乙醇等醇类,THF等醚类,或者它们的混合溶剂。
反应温度为-20℃~150℃、优选为0℃~100℃。
反应时间根据反应温度等而不同,通常为30分钟~48小时。
工序4
本工序是将亚氨酸酯转化为脒的工序,是使氨或铵盐反应、从而得到脒化合物6的工序。
作为本反应中使用的铵盐,可例举乙酸铵、氯化铵等。所使用的铵盐或氨优选以相对于亚氨酸酯为1~10倍摩尔当量的量使用。
本反应中可以根据需要在碱的存在下进行反应。作为所使用的碱,可例举例如三乙胺(以下称为“TEA”)、二异丙基乙胺(以下称为“DIPEA”)等有机碱。
作为所使用的溶剂,只要不参与反应就没有特别限定,通常使用甲醇、乙醇等醇类。
反应温度为-20℃~150℃、优选为0℃~100℃。
反应时间根据反应温度等而不同,通常为30分钟~48小时。
工序5
本工序是使脒化合物6和草乙酸二乙酯7或其盐在碱的存在下在合适的溶剂中反应、从而得到嘧啶化合物8的工序。
作为所使用的碱,可例举氢氧化钠、氢氧化钾、碳酸钠等无机碱,甲醇钠、乙醇钠等醇盐类。
所使用的碱优选以相对于脒化合物6为1~50倍摩尔当量的量使用。
作为本反应中使用的溶剂,只要不参与反应就没有特别限定,可使用例如THF、二甲氧基乙烷(以下称为“DME”)等醚类,甲醇、乙醇等醇类,水,或者它们的混合溶剂。
本反应中,反应温度为0℃~200℃、优选为0℃~100℃。
反应时间根据反应温度等而不同,通常为30分钟~24小时。
工序6
本工序是使羧酸8与胺化合物9在合适的溶剂中缩合得到化合物10的反应。或者,通过使羧酸8的反应性化合物与化合物9反应,也能制造化合物10。
作为羧酸8的反应性化合物,可例举例如酰卤(例如酰氯)、混合酸酐、咪唑阴离子、活性酰胺等酰胺缩合形成反应中通常使用的物质。
使用羧酸8的情况下,作为缩合剂,可使用例如1,1’-羰基二咪唑(以下称为“CDI”)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(以下称为“EDCI”)、O-(7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸酯(以下称为“HATU”)、叠氮磷酸二苯酯。
本反应中,缩合剂的用量合适的是相对于羧酸8为1~3倍摩尔当量。
本反应中可以根据需要在碱的存在下进行反应。作为所使用的碱,可例举例如TEA、DIPEA、吡啶等有机碱。
作为所使用的溶剂,只要不参与反应就没有特别限定,可使用例如THF、DME等醚类,二甲基甲酰胺(以下称为“DMF”)、N-甲基吡咯烷酮(以下称为“NMP”)等酰胺类,乙腈、丙腈等腈类,或者它们的混合溶剂。
本反应中,反应温度为-78℃~200℃、优选为-20℃~50℃。
反应时间根据反应温度等而不同,通常为10分钟~24小时。
工序7
本工序是使化合物10与磺酰氯11在合适的溶剂中反应、从而得到化合物12的工序。
作为所使用的磺酰氯11,可例举甲磺酰氯、对甲苯磺酰氯、苯磺酰氯等。
所使用的磺酰氯11优选以相对于化合物10为1~3倍摩尔当量的量使用。
本反应中可以根据需要在碱的存在下进行反应。作为所使用的碱,可例举例如TEA、DIPEA、吡啶等有机碱。
作为本反应中使用的溶剂,只要不参与反应就没有特别限定,可使用例如THF、DME等醚类,乙腈、丙腈等腈类,DMF、NMP等酰胺类,或者它们的混合溶剂。
本反应中,反应温度为-20℃~200℃、优选为0℃~100℃。反应时间根据反应温度等而不同,通常为30分钟~24小时。
工序8
本工序是使化合物12与胺化合物13在合适的溶剂中反应、从而得到化合物14的工序。
本反应中,胺化合物13的用量合适的是相对于化合物12为1~10倍摩尔当量。
本反应中可以根据需要在碱的存在下进行反应。作为所使用的碱,可例举例如TEA、DIPEA、吡啶等有机碱,氢氧化钠、碳酸氢钠、碳酸钠等无机碱。
作为本反应中使用的溶剂,只要不参与反应就没有特别限定,可使用例如THF、DME等醚类,乙腈、丙腈等腈类,DMF、NMP等酰胺类,乙醇、异丙醇等醇类,或者它们的混合溶剂。
本反应中,反应温度为0℃~200℃、优选为20℃~150℃。可以根据需要利用微波或者在密封条件下进行。
反应时间根据反应温度等而不同,通常为30分钟~24小时。
本发明化合物为甲苯磺酸盐一水合物的情况下,可通过在游离碱的溶液中添加对甲苯磺酸一水合物而获得。
本发明化合物如下述试验例所示,具有JAK1抑制活性。此外,本发明化合物因为具有JAK1抑制活性,所以具有抗炎症作用、免疫抑制作用、增殖抑制作用等。
因此,本发明化合物可以用作例如JAK1相关的疾病、可通过抗炎症作用、免疫抑制作用、增殖抑制作用等而期待有效性的疾病的预防剂或治疗剂。
作为可以应用本发明化合物的具体疾病,可例举自身免疫性疾病(例如类风湿性关节炎、牛皮癣关节炎、幼年型关节炎、卡斯特曼病、全身性红斑狼疮、斯耶格伦综合征、多发性硬化症、炎性肠病、贝切特病、重症肌无力、I型糖尿病、免疫球蛋白肾病、自身免疫性甲状腺病、银屑病、硬皮病、狼疮性肾炎、干眼症、血管炎(例如大动脉炎、巨细胞动脉炎、显微镜下多血管炎、肉芽肿性多血管炎、嗜酸性肉芽肿性多血管炎)、皮肤肌炎、多肌炎、视神经脊髓炎等)、炎症性疾病(例如特应性皮炎、接触性皮炎、湿疹、瘙痒症、食物过敏、支气管哮喘、嗜酸性粒细胞性肺炎、慢性阻塞性肺病、过敏性鼻炎、慢性鼻窦炎、嗜酸性粒细胞性鼻窦炎、鼻息肉、过敏性结膜炎、骨关节炎、强直性脊柱炎、川崎病、伯格氏病、结节性多动脉炎、IgA血管炎等)、增殖性疾病(例如实体癌、血液癌、淋巴系统恶性肿瘤、骨髓增殖性疾病、多发性骨髓瘤、肺纤维化、嗜酸性粒细胞增多症等)、突发性聋、糖尿病肾病、斑秃、骨髓移植排异或脏器移植排异等。
将本发明化合物作为医药进行给药的情况下,可以将本发明化合物直接或作为在药学上可接受的无毒性且惰性的载体中含有例如0.001%~99.5%、优选0.1%~90%的医药组合物,对包括人在内的哺乳动物给药。
作为载体,可例举固体、半固体或液状的稀释剂、填充剂、及其它的处方用助剂的一种以上。本发明的医药组合物较好是以给药单位形态进行给药。医药组合物可以组织内给药、口服给药、静脉内给药、局部给药(经皮给药、滴眼、腹腔内、胸腔内等)或者以经过直肠的方式给药。当然,给药是以适合于这些给药方法的剂型来进行的。
作为医药的用量较好是在考虑到年龄、体重、疾病的种类、程度等患者的状态、给药途径、本发明化合物的种类等的基础上进行调整,通常对于成人而言,以本发明化合物的有效成分量计,在口服给药的情况下,1天的用量合适的是在0.01mg~5g/成人的范围内,优选在1mg~500mg/成人的范围内。根据情况,有时该范围以下的量即可,有时反而需要该范围以上的用量。通常,1天给药1次或分数次给药,或在静脉内给药的情况下,可以进行急速给药或者在例如24小时以内进行持续给药。
实施例
以下,举出实施例、试验例及制剂例对本发明进行更详细的说明,但本发明并不限定于此。
粉末X射线衍射图采用SmartLab(理学株式会社((株)リガク)制)(靶:Cu,电压:45kV,电流:200mA,扫描速度:47.3度/分钟)测定。
质谱通过高效液相色谱质谱测定。作为电离法,采用电喷雾电离法。观测到的质谱的值以m/z表示。
高效液相色谱质谱的测定条件如下所述。
分析仪器:ACQUITY UPLC MS/PDA系统(沃特世公司(ウォーターズ社)制)
质谱仪:Waters 3100MS检测器
光二极管阵列检测器:ACQUITY PDA检测器(UV检测波长:210~400nm)
柱:Acquity BEH C18,1.7μm、2.1×50mm
流速:0.5mL/分钟
柱温:40℃
溶剂:
A液:0.1%甲酸/H2O(v/v;下同)
B液:0.1%甲酸/乙腈
采用以下所示的高效液相色谱条件来确定光学纯度。
分析仪器:SHIMADZU LC-10AS(岛津制作所(島津製作所)制)
检测器:SPD-10A(岛津制作所制,UV检测波长:254nm)
柱:Chiralcel AD-HΦ4.6mm×250mm(大赛璐(ダイセル)制)
流速:1mL/分钟
柱温:40℃
溶剂:己烷/乙醇/二乙胺=850/150/1(v/v/v)
实施例中,使用以下的缩写。
DMF:二甲基甲酰胺
DMSO:二甲亚砜
DIPEA:N,N-二异丙基乙胺
TEA:三乙胺
THF:四氢呋喃
TFA:三氟乙酸
NMP:N-甲基吡咯烷酮
HATU:O-(7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸酯
MS:质谱
LCMS:高效液相色谱质谱
ESI:电喷雾电离法(Electron Spray Ionization)
M:摩尔浓度
v/v:容量/容量
参考例1吡唑并[5,1-b][1,3]噻唑-7-甲腈
[工序1]2-(噻唑-2-基)乙腈的制造
向氰基乙酸叔丁酯(28g)的DMF(100mL)溶液中在冰冷下少量逐次加入60%氢化钠(7.9g),搅拌10分钟。向其中加入2-溴噻唑(25g),在室温下搅拌15分钟,接着在120℃下搅拌2小时。向反应混合液中加入1M盐酸水溶液后,用乙酸乙酯萃取。用水洗涤有机层后,用无水硫酸镁干燥,进行减压浓缩。将所得残渣用己烷洗涤,悬浮于甲苯(200mL),加入对甲苯磺酸一水合物(2.0g),在105℃下搅拌2小时。将反应液用乙酸乙酯稀释后,用饱和碳酸氢钠水溶液洗涤。将所得有机层用无水硫酸镁干燥后,进行减压浓缩。所得残渣用硅胶柱色谱纯化,得到标题化合物(7.0g)。
MS(m/z):125[M+H]+
[工序2]吡唑并[5,1-b][1,3]噻唑-7-甲腈的制造
向工序1中得到的2-(噻唑-2-基)乙腈(5g)的二氯甲烷(50mL)溶液中在冰冷下加入O-(基磺酰基)羟胺(例如按照Organic Process Research&Development,2009,13,263-267.中记载的方法合成)的二氯甲烷(20mL)溶液,在室温下搅拌2小时。在冰冷下向反应混合物中加入二乙醚,滤取析出的固体物。将所得固体物悬浮于原甲酸三乙酯(35mL),在120℃下搅拌1小时。将反应溶液减压浓缩,所得残渣用硅胶柱色谱纯化,得到标题化合物(2.5g)。
MS(m/z):150[M+H]+
参考例2 6-羟基-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸
向参考例1中得到的吡唑并[5,1-b][1,3]噻唑-7-甲腈(6g)的甲醇(150mL)溶液中加入28%甲醇钠甲醇溶液(24.6mL),在室温下搅拌3小时。接着,加入氯化铵(12.9g),在90℃下搅拌1小时。将反应液减压浓缩,向所得残渣中加入草酰乙酸二乙酯钠(33.8g)的5M氢氧化钠水溶液(200mL),在100℃下搅拌过夜。向反应混合液中加入浓盐酸使其呈酸性,滤取析出的固体物。将所得固体物溶解于5M氢氧化钾水溶液,用氯仿洗涤。向水层中加入浓盐酸使其呈酸性,滤取析出的固体物,干燥,得到标题化合物(10g)。
MS(m/z):263[M+H]+
参考例3 6-氯-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸甲酯
将参考例2中得到的6-羟基-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸(1.4g)悬浮于磷酰氯(20mL),加入二乙基苯胺(1.6g),在130℃下搅拌2小时。将反应溶液减压浓缩,在冰冷下向反应混合物中加入甲醇(100mL),搅拌10分钟。将反应液用氯仿稀释后,用水洗涤。将所得有机层用无水硫酸镁干燥后,进行减压浓缩。所得残渣用硅胶柱色谱纯化,得到标题化合物(910mg)。
MS(m/z):297[M+H]+
参考例4(1-{[6-氯-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸甲酯
将参考例2中得到的6-羟基-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸(820mg)悬浮于磷酰氯(5.0mL),加入二乙基苯胺(0.47g),在110℃下搅拌2小时。将反应溶液减压浓缩,在冰冷下溶解于二氯甲烷(40mL),加入DIPEA(5.4mL)和工序1中得到的哌啶-4-基氨基甲酸甲酯(594mg),在室温下搅拌30分钟。将反应液用氯仿稀释后,用饱和碳酸氢钠水溶液洗涤。将所得有机层用无水硫酸镁干燥后,进行减压浓缩。所得残渣用硅胶柱色谱纯化,得到标题化合物(910mg)。
MS(m/z):421、423[M+H]+
参考例5 N-(1-{[6-氯-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)环丙烷羧酰胺
按照参考例4的方法,使用参考例2中得到的6-羟基-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸(8g),用N-(哌啶-4-基)环丙烷羧酰胺(例如按照Jornal of Medicinal Chemistry,2010,53,6386-6397.中记载的方法合成,5.65g)来代替哌啶-4-基氨基甲酸甲酯进行合成,得到标题化合物(6.65g)。
MS(m/z):431、433[M+H]+
参考例6 6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸
[工序1]6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸甲酯的制造
向参考例3中得到的6-氯-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸甲酯(1.0g)的DMF(10mL)溶液中加入DIPEA(1.8mL)、(1S)-1-环丙基乙胺(320mg),在80℃下搅拌3小时。用硅胶柱色谱纯化反应混合物,得到标题化合物(1.1g)。
MS(m/z):344[M+H]+
[工序2]6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸的制造
向工序1中得到的6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸甲酯(600mg)的THF(15mL)、水(5mL)溶液中加入氢氧化锂一水合物(100mg),在室温下搅拌1小时。向反应混合液中加入1M盐酸水溶液使其呈酸性后,减压蒸除THF,用氯仿萃取。将所得有机层用无水硫酸钠干燥,进行减压浓缩,得到标题化合物(470mg)。
MS(m/z):330[M+H]+
参考例7 3-氨基-4-羟基-1,3-噻唑烷-2-硫酮
向THF(750mL)中加入硼氢化钠(19.1g),在5℃以下分批添加N-氨基绕丹宁(250g)的THF(500mL)浆料。在5℃以下搅拌30分钟后,滴加甲醇(111mL),搅拌2小时。将浓盐酸(44mL)用水(500mL)稀释后滴加,然后滴加水(1000mL),在10℃以下搅拌1小时。将析出的晶体过滤,用水(600mL)洗涤后,在减压下在40℃下干燥,得到标题化合物(209.1g)。
MS(m/z):151[M+H]+
参考例8 2-氯-2-氰基乙烯-1-油酸钠
在20℃以下向甲醇钠(14.3g)的环戊基甲基醚(300mL)浆料中滴加甲酸甲酯(17.5g)后,滴加氯乙腈(20g),在30℃以下搅拌3小时。反应结束后,将析出的晶体过滤,用环戊基甲基醚(40mL)洗涤后,在减压下在40℃下干燥,得到标题化合物(29.2g)。
参考例9吡唑并[5,1-b][1,3]噻唑-7-羧基亚氨酸甲酯盐酸盐
向3-氨基-4-羟基-1,3-噻唑烷-2-硫酮(50g)的2-丙醇(250mL)浆料中添加浓硫酸(48.97g),在80℃下加热搅拌1小时。进行冷却,在40℃以下添加乙腈(500mL)、参考例8中得到的2-氯-2-氰基乙烯-1-油酸钠(62.65g),在80℃下搅拌4小时。进行冷却,添加活性炭(10g),在室温下搅拌30分钟。过滤不溶物,用乙腈洗涤3次(100mL)。将滤液减压浓缩,接着用甲醇(100mL)共沸3次以除去乙腈,然后向浓缩物中加入甲醇(150mL)和THF(150mL)。将其添加至在20℃以下由甲醇(350mL)和乙酰氯(209g)制备的溶液中。在室温下搅拌过夜后,添加THF(350mL),进一步在10℃以下搅拌1小时。将析出的晶体过滤,用THF(300mL)洗涤后,在减压下在50℃下干燥,得到标题化合物(45.5g)。
MS(m/z):182[M+H]+
参考例10吡唑并[5,1-b][1,3]噻唑-7-羧基酰亚胺酰胺乙酸盐
向吡唑并[5,1-b][1,3]噻唑-7-羧基亚氨酸甲酯盐酸盐(200g)的甲醇(1000mL)浆料中添加乙酸铵(85.15g)后,添加DIPEA(142.82g),在65℃下搅拌1小时。反应结束后,进行冷却,在室温下滴加乙腈(2000mL),在10℃以下搅拌1小时。将析出的晶体过滤,用乙腈(400mL)洗涤后,在减压下在50℃下干燥,得到标题化合物(185.12g)。
元素分析值(以C6H6N4S·C2H4O2计)
计算值(%)C:42.47,H:4.46,N:24.76
实测值(%)C:42.18,H:4.25,N:24.41
参考例11 6-羟基-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸
在10℃以下向氢氧化钠(39.08g)的水溶液(900mL)中添加草乙酸二乙酯钠(130.65g),搅拌1小时。向其中添加吡唑并[5,1-b][1,3]噻唑-7-羧基酰亚胺酰胺乙酸盐(90g),在50℃下加热搅拌3小时。然后进行冷却,在30℃以下加入浓盐酸(138g),确认液性已达pH1~2后,在室温下搅拌过夜。滤取析出的晶体,用水(360mL)洗涤后,在60℃下干燥,得到标题化合物(107.17g)。
MS(m/z):263[M+H]+
参考例12{1-[6-羟基-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羰基]哌啶-4-基}氨基甲酸甲酯
向6-羟基-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸(238g)的DMF(714mL)浆料中添加TEA(275.51g),在50℃下搅拌30分钟。进行冷却后,在20℃以下添加1,1’-羰基二咪唑(323.76g)。搅拌30分钟后,添加哌啶-4-基氨基甲酸甲酯甲苯磺酸盐(449.79g),搅拌30分钟。反应结束后,在室温下滴加乙腈(3570mL),搅拌过夜。将析出的晶体过滤,用乙腈(480mL)洗涤后,在减压下在60℃下干燥,得到标题化合物(377.79g)。
MS(m/z):403[M+H]+
参考例13 6-{4-[(甲氧基羰基)氨基]哌啶-1-羰基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基4-甲基苯-1-磺酸酯
向{1-[6-羟基-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羰基]哌啶-4-基}氨基甲酸甲酯(365g)的乙腈(1825mL)浆料中添加TEA(275.33g)后,添加4-甲苯磺酰氯(259.36g),在50℃下加热搅拌1小时。反应结束后,进行冷却,在室温下滴加水(3650mL),在10℃以下搅拌1小时。将析出的晶体过滤,用水(730mL)洗涤后,在减压下在60℃下干燥,得到标题化合物(442.08g)。
MS(m/z):557[M+H]+
实施例1[1-({6-[(2S)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯甲苯磺酸盐一水合物
[工序1][1-({6-[(2S)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯的制造
向参考例13中得到的6-{4-[(甲氧基羰基)氨基]哌啶-1-羰基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基4-甲基苯-1-磺酸酯(1.0g)的乙腈(7.0mL)溶液中添加DIPEA(0.67g)、(2S)-丁烷-2-胺(0.4g)后,封管,在100℃下加热搅拌2小时。反应结束后,将反应液用乙酸乙酯稀释后,用水(30mL)和饱和食盐水(30mL)洗涤。将有机层用无水硫酸镁干燥,进行减压浓缩,然后用柱色谱纯化,得到标题化合物(650mg)。通过高效液相色谱确认,所得标题化合物的光学纯度在99%以上(保留时间:35.7分钟)。
MS(m/z):458[M+H]+
[工序2][1-({6-[(2S)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯甲苯磺酸盐一水合物的制造
向实施例1工序1中得到的[1-({6-[(2S)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯(202mg)中加入乙腈(5mL),加热至50℃。加入对甲苯磺酸一水合物(83mg),在室温下搅拌过夜。滤取析出的晶体,进行减压干燥,得到标题化合物的晶体(210mg)。粉末X射线衍射图示于图1。元素分析显示,所得晶体为一水合物。
元素分析值(以C21H27N7O3S·C7H8O3S·1.0H2O计)
计算值(%)C:51.92,H:5.76,N:15.14
实测值(%)C:51.54,H:5.92,N:15.03
实施例2[1-({6-[(2R)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯甲苯磺酸盐一水合物
[工序1][1-({6-[(2R)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯的制造
向参考例13中得到的6-{4-[(甲氧基羰基)氨基]哌啶-1-羰基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基4-甲基苯-1-磺酸酯(1.5g)的乙腈(10mL)溶液中添加DIPEA(1.0g)、(2R)-丁烷-2-胺(0.59g)后,封管,在100℃下加热搅拌2小时。反应结束后,将反应液用乙酸乙酯稀释后,用水(50mL)和饱和食盐水(30mL)洗涤。将有机层用无水硫酸镁干燥,进行减压浓缩,然后用柱色谱纯化,得到标题化合物(0.93g)。通过高效液相色谱确认,所得标题化合物的光学纯度在99%以上(保留时间:29.1分钟)。
MS(m/z):458[M+H]+
[工序2][1-({6-[(2R)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯甲苯磺酸盐一水合物的制造
向实施例2工序1中得到的[1-({6-[(2R)-丁烷-2-基氨基]-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基}羰基)哌啶-4-基]氨基甲酸甲酯(140mg)中加入乙腈(3.5mL),加热至50℃。加入对甲苯磺酸一水合物(58mg),在室温下搅拌过夜。滤取析出的晶体,进行减压干燥,得到标题化合物的晶体(152mg)。粉末X射线衍射图示于图2。元素分析显示,所得晶体为一水合物。
元素分析值(以C21H27N7O3S·C7H8O3S·1.0H2O计)
计算值(%)C:51.92,H:5.76,N:15.14
实测值(%)C:51.72,H:5.84,N:15.14
实施例3(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸甲酯甲苯磺酸盐一水合物
[工序1](1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸甲酯的制造
向参考例13中得到的6-{4-[(甲氧基羰基)氨基]哌啶-1-羰基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基4-甲基苯-1-磺酸酯(1.0g)的乙腈(7.0mL)溶液中添加DIPEA(0.69g)、(1S)-1-环丙基乙胺(460mg)后,封管,在100℃下加热搅拌2小时。反应结束后,将反应液用乙酸乙酯稀释后,用水(50mL)和饱和食盐水(30mL)洗涤。将有机层用无水硫酸镁干燥,进行减压浓缩,然后用柱色谱纯化,得到标题化合物(720mg)。
[工序2](1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸甲酯甲苯磺酸盐一水合物
向实施例3工序1中得到的(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸甲酯(201mg)中加入乙腈(5mL),加热至50℃。加入对甲苯磺酸一水合物(81mg),在室温下搅拌过夜。滤取析出的晶体,进行减压干燥,得到标题化合物的晶体(237mg)。粉末X射线衍射图示于图3。元素分析显示,所得晶体为一水合物。
元素分析值(以C22H27N7O3S·C7H8O3S·1.0H2O计)
计算值(%)C:52.79,H:5.65,N:14.86
实测值(%)C:52.72,H:5.54,N:14.82
比旋光度[α]D25=-44.4(c=1.00、DMSO)
实施例4(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)氨基甲酸乙酯
向参考例6中得到的6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸(640mg)的DMF(5.0mL)溶液中加入哌啶-4-基氨基甲酸乙酯(例如按照US1990/4918073中记载的方法合成)(310mg)、DIPEA(730μL)、HATU(1.1g),在室温下搅拌1小时。用硅胶柱色谱纯化反应混合物,得到标题化合物(410mg)。向所得标题化合物(350mg)中加入乙酸乙酯(7mL),加热溶解,在室温下搅拌20小时。滤取析出的晶体,进行减压干燥,得到标题化合物的晶体(280mg)。粉末X射线衍射图示于图4。
MS(m/z):484[M+H]+
元素分析值(以C23H29N7O3S计)
计算值(%)C:57.12,H:6.04,N:20.27
实测值(%)C:56.81,H:6.12,N:20.28
比旋光度[α]D25=-44.2(c=1.00、DMSO)
实施例5 N-(1-{[6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)环丙烷羧酰胺
向参考例6中得到的6-{[(1S)-1-环丙基乙基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-羧酸(350mg)的DMF(8.0mL)溶液中加入N-(哌啶-4-基)环丙烷羧酰胺(268mg)、DIPEA(552μL)、HATU(606mg),在室温下搅拌1小时。用硅胶柱色谱纯化反应混合物,得到标题化合物(410mg)。向所得标题化合物(350mg)中加入乙酸乙酯(7mL),加热溶解,在室温下搅拌20小时。滤取析出的晶体,进行减压干燥,得到标题化合物的晶体(280mg)。粉末X射线衍射图示于图5。
元素分析值(以C24H29N7O2S计)
计算值(%)C:60.12,H:6.09,N:20.44
实测值(%)C:59.89,H:6.37,N:20.22
MS(m/z):480[M+H]+
比旋光度[α]D25=-45.2(c=1.00、DMSO)
实施例6 N-(1-{[6-{[(2R)-3,3-二甲基丁烷-2-基]氨基}-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)环丙烷羧酰胺
在氩气氛下向参考例5中得到的N-(1-{[6-氯-2-(吡唑并[5,1-b][1,3]噻唑-7-基)嘧啶-4-基]羰基}哌啶-4-基)环丙烷羧酰胺(550mg)的叔丁醇(20mL)溶液中加入TEA(534μL)、(2R)-3,3-二甲基丁烷-2-胺(258mg),在90℃下搅拌过夜。将反应混合物减压浓缩,所得残渣用硅胶柱色谱纯化,得到标题化合物(570mg)。向所得标题化合物(100mg)中加入乙酸乙酯(2mL),加热溶解,在室温下搅拌20小时。滤取析出的晶体,进行减压干燥,得到标题化合物的晶体(70mg)。粉末X射线衍射图示于图6。
元素分析值(以C25H33N7O2S计)
计算值(%)C:60.58,H:6.71,N:19.78
实测值(%)C:60.20,H:6.96,N:19.53
MS(m/z):496[M+H]+
比旋光度[α]D25=+14.0(c=1.00、DMSO)
试验例1 JAK酪氨酸激酶抑制作用
1.受试物质的制备
受试物质用二甲亚砜(DMSO)调配至10mM,进一步用DMSO分别稀释至1000、100、10、1、0.1、0.01μM的浓度。JAK1使用10mM、1000μM、100μM、10μM、1μM、0.1μM这6种浓度的DMSO溶液,JAK2和JAK3使用1000μM、100μM、10μM、1μM、0.1μM、0.01μM这6种浓度的DMSO溶液,用分析缓冲液稀释20倍,配制成受试物质溶液。作为阴性对照,使用用分析缓冲液将DMSO稀释20倍而得的溶液。作为分析缓冲液,使用15mM的Tris-HCl(pH7.5)、0.01(v/v)%的吐温-20(Tween-20)、1mM的二硫苏糖醇。
2.1mM ATP下的JAK酪氨酸激酶抑制活性的测定
活性的测定采用ELISA法。以每10μL的量(n=2)将受试物质溶液添加到涂有链霉亲和素的96孔板(DELFIA条形板(DELFIA Strip Plate)8×12孔、珀金埃尔默公司(PerkinElmer社)),以每20μL的量添加底物溶液(生物素标记肽底物1250nM(JAK1)、625nM(JAK2、JAK3)、2.5mM的ATP(终浓度1mM)、25mM的MgCl2、15mM的Tris-HCl(pH7.5)、0.01(v/v)%的吐温-20、1mM的二硫苏糖醇),进行搅拌。最后,以每20μL的量添加JAK酪氨酸激酶(卡纳生物科学公司(カルナバイオサイエンス社))(已用分析缓冲液稀释至7.5nM(JAK1)、0.75nM(JAK2、JAK3)),进行搅拌,在30℃下进行1小时的反应。用清洗缓冲液(50mM的Tris-HCl(pH7.5)、150mM的NaCl、0.02(v/v)%的吐温-20)将板清洗4次后,以每150μL的量添加封闭缓冲液(0.1%的牛血清蛋白(Bovine Serum Albumin)、50mM的Tris-HCl(pH7.5)、150mM的NaCl、0.02(v/v)%的吐温-20),在30℃下进行1小时的封闭。除去封闭缓冲液,以每100μL的量添加辣根过氧化物酶(Horse radish Peroxidase)标记抗磷酸酪氨酸抗体(BD生物科学公司(BD Bioscience社))(已用封闭缓冲液稀释至10000倍),在30℃下孵育30分钟。用清洗缓冲液将板清洗4次,以每100μL的量添加3,3’,5,5’-四甲基联苯胺溶液(半井泰斯科公司(ナカライテスク社)),显色10分钟。以每100μL的量添加0.1M硫酸,使反应停止。用酶标仪(BIO-RAD公司)测定450nm处的吸光度。
3.测定结果的分析
对于测得的吸光度,利用SAS系统(赛仕软件研究所公司(SAS Institute Inc.))进行非线性回归分析,算出将各酪氨酸激酶活性抑制50%的受试物质的浓度(IC50)。其结果示于以下的表1。
[表1]
将上述试验例1的实施例化合物作为受试物质,进行以下的试验(试验例2、试验例3和试验例4)。
试验例2曲霉诱发气道炎症模型中的抑制作用
将烟曲霉(Aspergillus fumigatus)提取物(格里尔实验室公司(Greer laboratories社))用PBS溶液调配至400μg/mL。在第0天、第1天、第7天、第8天将调配的烟曲霉溶液50μL对小鼠滴鼻给药。滴鼻给药在早晨的受试物质给药的1小时后进行。受试物质给药从第0天进行至第9天,每天进行早晚2次。使受试物质以10mg/mL的浓度悬浮于0.5%甲基纤维素,以10mL/kg的用量口服给药。在第10天回收支气管肺胞灌洗液(bronchoalveolar lavage fluid:BALF),用Celltac(日本光电株式会社(日本光電社))测定BALF中的总白细胞数。接着,用ADVIA 120(西门子医学诊断公司(Siemens Healthcare Diagnostics社))算出总白细胞数中的嗜酸性粒细胞数的比例,乘以总白细胞数,从而算出BALF中的嗜酸性粒细胞数。将烟曲霉提取物处理+0.5%甲基纤维素给药组的嗜酸性粒细胞数设为抑制率0%,将烟曲霉提取物未处理+0.5%甲基纤维素给药组的嗜酸性粒细胞数设为抑制率100%,算出各受试物质的抑制率。
试验例3由IL-4刺激导致的STAT6磷酸化的抑制作用
1.受试物质的制备
将受试物质用二甲亚砜(DMSO)调配至10mM,进一步用DMSO稀释至300、100μM的浓度。再用RPMI1640培养基稀释100倍,将稀释后的溶液作为受试物质溶液。作为阴性对照,使用用RPMI1640将DMSO稀释100倍而得的溶液。
2.磷酸化STAT6活性的测定
将50μL的受试物质溶液或阴性对照溶液与400μL的DND39细胞液(细胞数:105个细胞)混合,在37℃下振荡30分钟。添加50μL的白细胞介素-4(10ng/mL)作为刺激物质,振荡15分钟。添加500μL的固定试剂Fixation buffer(BD生物科学公司),振荡10分钟,使反应停止。离心除去上清后,添加500μL的膜透化试剂Perm buffer III(BD生物科学公司),在4℃下孵育30分钟。用染色缓冲液(Stain buffer)(BD生物科学公司)进行2次清洗操作后,添加Alexa Fluor 647小鼠抗Stat6(pY641)(BD生物科学公司),在冷暗处孵育30分钟。将所得细胞溶液供至流式细胞仪。将白细胞介素-4刺激阴性对照组荧光强度的GEOMEAN值设为抑制率0%,将无刺激阴性对照组荧光强度的GEOMEAN值设为抑制率100%,算出各受试物质的抑制率,确认这些受试物质抑制IL-4的信号。
试验例4由IL-7刺激导致的STAT5磷酸化的抑制作用
1.受试物质的制备
将受试物质用二甲亚砜(DMSO)调配至10mM。再用RPMI1640培养基稀释100倍,将稀释后的溶液作为受试物质溶液。作为阴性对照,使用用RPMI1640将DMSO稀释100倍而得的溶液。
2.磷酸化STAT5活性的测定
向人新鲜血100μL中加入10μL的受试物质溶液或阴性对照溶液,在37℃下振荡30分钟。以每10μL的量添加白细胞介素-7(100ng/mL)作为刺激物质,振荡15分钟。将裂解/固定缓冲液(Lyse/fix buffer)(BD生物科学公司)用蒸馏水稀释5倍,添加1.4mL。然后,振荡10分钟后,进行离心,将细胞分离。除去上清后,添加1mL的PBS。离心除去PBS后,添加500μL的Perm buffer III(BD生物科学公司),在4℃下孵育30分钟。用染色缓冲液Stain buffer(BD生物科学公司)进行2次清洗操作后,添加Alexa Fluor 647小鼠抗Stat5抗体(pY694)(BD生物科学公司),在冷暗处孵育30分钟。将所得细胞溶液供至流式细胞仪。将IL-7刺激阴性对照组荧光强度的GEOMEAN值设为抑制率0%,将无刺激阴性对照组荧光强度的GEOMEAN值设为抑制率100%,算出各受试物质的抑制率,确认这些受试物质抑制IL-7的信号。
如试验例1~试验例4所述,本发明化合物显示出JAK1抑制活性,对体内炎症模型有效。
产业上利用的可能性
本发明化合物显示出JAK1抑制活性,因此可以用作自身免疫性疾病(例如类风湿性关节炎、牛皮癣关节炎、幼年型关节炎、卡斯特曼病、全身性红斑狼疮、斯耶格伦综合征、多发性硬化症、炎性肠病、贝切特病、重症肌无力、I型糖尿病、免疫球蛋白肾病、自身免疫性甲状腺病、银屑病、硬皮病、狼疮性肾炎、干眼症、血管炎(例如大动脉炎、巨细胞动脉炎、显微镜下多血管炎、肉芽肿性多血管炎、嗜酸性肉芽肿性多血管炎)、皮肤肌炎、多肌炎、视神经脊髓炎等)、炎症性疾病(特应性皮炎、接触性皮炎、湿疹、瘙痒症、食物过敏、支气管哮喘、嗜酸性粒细胞性肺炎、慢性阻塞性肺病、过敏性鼻炎、慢性鼻窦炎、嗜酸性粒细胞性鼻窦炎、鼻息肉、过敏性结膜炎、骨关节炎、强直性脊柱炎、川崎病、伯格氏病、结节性多动脉炎、IgA血管炎等)、增殖性疾病(例如实体癌、血液癌、淋巴系统恶性肿瘤、骨髓增殖性疾病、多发性骨髓瘤、肺纤维化、嗜酸性粒细胞增多症等)、突发性聋、糖尿病肾病、斑秃、骨髓移植排异或脏器移植排异等的治疗剂。
制剂例1
片剂(内服片)
处方1片80mg中
实施例1的本发明化合物 5.0mg
玉米淀粉 46.6mg
结晶纤维素 24.0mg
甲基纤维素 4.0mg
硬脂酸镁 0.4mg
用常规的方法将该比例的混合粉末压片成形以制成内服片。
- 还没有人留言评论。精彩留言会获得点赞!