用于高乙烯基嵌段共聚的极性改性剂体系的制作方法
本申请要求于2016年9月9日申请的美国临时专利申请序列号62/385,776和于2017年1月31日申请的美国临时专利申请序列号62/452,406的优先权,在此通过引用并入本文。
背景技术:
:1.
技术领域:
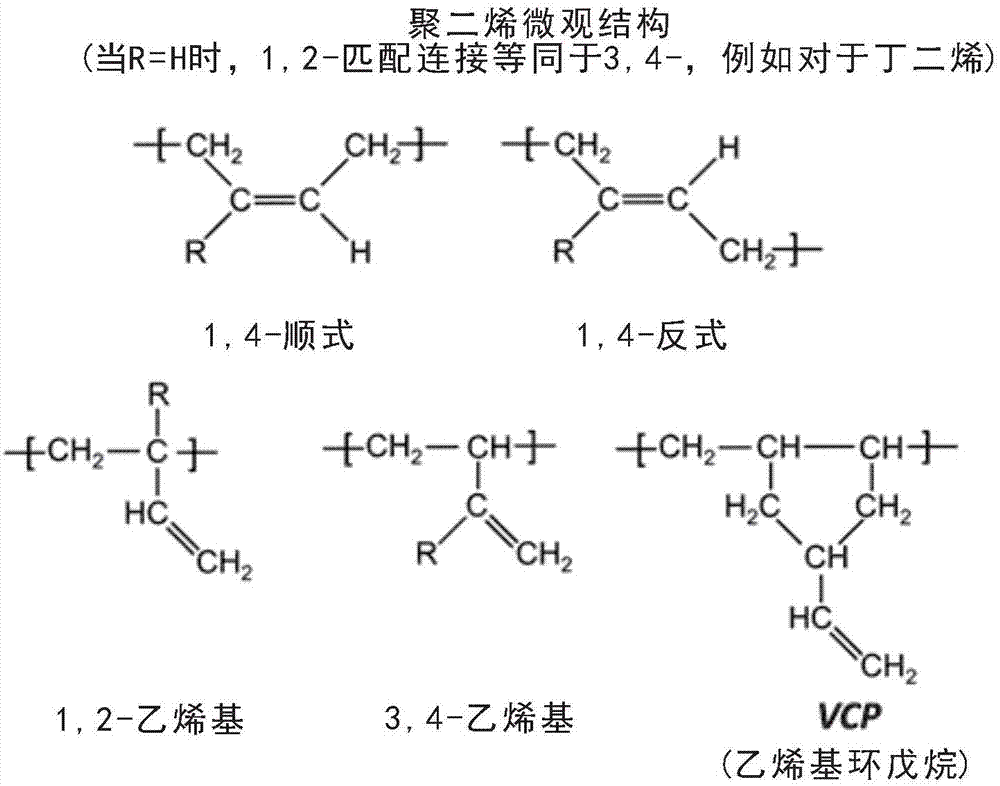
本发明涉及通过在脂族烃溶液中有机锂引发的阴离子聚合合成共轭二烯单体和乙烯基芳族单体的嵌段共聚物的极性改性剂和方法。更具体地,用本发明的极性改性剂体系和方法生产的嵌段共聚物有利地结合了非常高水平的具有侧接乙烯基双键的重复单元与低水平的乙烯基环戊烷重复单元。此外,本发明使得能够以快速聚合速率生产这种嵌段共聚物,其具有窄分子量分布,在共聚单体嵌段之间具有低含量的无规化共聚单体重复单元转变,并且采用严苛度低于现有技术的冷却要求来进行聚合。2.相关技术的说明由烷基锂引发的脂族烃溶液中的阴离子聚合的分批和半分批工艺是生产共轭二烯单体(如丁二烯和异戊二烯)和乙烯基芳族单体(如苯乙烯)的嵌段共聚物的通用技术。这是因为这些类型的单体在脂族烃中的阴离子聚合和共聚通过既缺少终止、也缺少链转移反应的链反应机制发生;诸如此类的聚合机理是所谓的活性聚合。聚合的活性允许多种嵌段序列构型、偶合方案、端基官能化化学反应以及对组成、分子量和分子量分布的精确控制。本发明的目的是提供新的极性改性剂体系,该体系被特别定向以产生独特种类的嵌段共聚物,该嵌段共聚物通过共轭二烯的1,2或3,4-加成掺入非常高水平的侧接乙烯基键。已公开了许多极性改性剂,其可影响朝这种微结构的共轭二烯加成模式,然而,非常常见的缺点是它们在聚合温度上升时促进1,2或3,4-加成的能力显著下降(hsie,h.l.;quirk,r.p.,anionicpolymerization:principlesandpracticalapplications,marceldekker,1996,第217页)。工业工艺的典型操作温度和有限的除热能力排除了极性改性剂获得基于共轭二烯高于80wt%的1,2-乙烯基含量的多数选择。此外,各种极性改性剂由于链转移反应而易于加宽分子量分布(hsie,h.l.;quirk,r.p.,anionicpolymerization:principlesandpracticalapplications,marceldekker,1996,第188-189和217页)。当以易于获得高乙烯基含量(>70%)的量使丁二烯在极性改性剂存在下聚合时,一部分丁二烯作为乙烯基环戊烷(vcp)的重复单元被掺入。需要两个丁二烯分子来获得单个vcp重复单元,并且产生单个侧接双键(参见图1)。luxton等(polymer,第22卷,第3期,1981年,第382-386页)解释了增加vcp含量的促成因素是:聚合期间的低丁二烯浓度、使用高装料的tmeda和/或钠抗衡离子,以及高聚合温度。vcp含量增加聚丁二烯链段的玻璃转化温度(tg),降低了主链的柔韧性和低温性质,这影响嵌段共聚物的弹性体热塑性组合物的热性质和动态性质。本发明克服了这些障碍,增强了生产率和对嵌段共聚物分子属性的控制。美国专利号7,851,558提供了采用极性改性剂(n,n,n’,n’-四甲基乙二胺(tmeda)、双四氢糠基丙烷(dthfp)、二哌啶基乙烷(dipip)和四氢呋喃(thf))进行的高乙烯基苯乙烯-丁二烯二嵌段和苯乙烯-丁二烯-苯乙烯三嵌段共聚物的烷基锂引发的阴离子聚合的实例。所报道的1,2-乙烯基异构体含量对于tmeda高达78%,对于dthfp高达84%,对于dipip高达96%,且对于thf高达54%。然而,聚合温度保持非常低(25℃),溶剂与单体的质量比相当高(约12),并且丁二烯嵌段在两小时的时间跨度内聚合。美国专利号5,906,956显示了作为叔戊醇钠(sta)的低分子量醇盐和tmeda的协同作用,这使得丁二烯在75℃下均聚成为可能,得到10分钟内的完全转化和83%的乙烯基掺入。sta与锂的摩尔比为至少0.5,并且tmeda与锂的摩尔比为至少2。没有显示合成共轭二烯和乙烯基芳族单体的嵌段共聚物的实验数据,也没有显示合成的聚丁二烯的vcp含量的实验数据。美国专利号6,140,434通过使用薄荷醇钠(smt)改善了tmeda/醇盐配方的工业操作可行性。smt优于sta的主要优点是从工业操作的角度来看,sta在汽提阶段与水接触时产生挥发性醇,该醇难以从溶剂再循环中去除并使阴离子聚合中毒,而smt因其高沸点和其薄荷醇副产物的高沸点而不影响溶剂再循环的纯度。显示了一个实例,即使用约8.09的溶剂与单体的比率、8/0.25/1的tmeda/smt/li摩尔比、65℃的反应温度和10分钟的反应时间,用85%的乙烯基掺入制备聚丁二烯。没有显示合成共轭二烯和乙烯基芳族单体的嵌段共聚物的实验数据。没有显示合成共轭二烯和乙烯基芳族单体的嵌段共聚物的实验数据,也没有显示合成的聚丁二烯的vcp含量的实验数据。美国专利号8,138,270显示tmeda/醇钠/正丁基锂极性改性剂/引发剂体系制备高乙烯基sbs型三嵌段共聚物。获得了具有窄重量分布、多分散性指数mw/mn为1.06的sbs共聚物,其总乙烯基水平基于丁二烯重量报告为76.8%,tmeda/叔戊醇钠/正丁基锂比率为1.8/0.055/1。在半连续模式温度下进行聚合,将丁二烯沿100分钟时段进给至反应器中,同时将反应温度控制在70℃。当用叔丁醇钠替代叔戊醇钠时,获得非常类似的结果。显示如果叔戊醇钠/li摩尔比增加至0.12,则发生分子量分布加宽至1.23的多分散性指数。未报道vcp含量。美国专利号7,740,760利用tmeda/smt体系通过升高vcp含量来增加聚丁二烯和无规苯乙烯-丁二烯共聚物(sbr)的tg。当评价在停留时间为40分钟且在80℃下操作的连续工艺反应器中使用每摩尔活性锂4.9摩尔tmeda进行的sbr合成时,共聚物展现10%vcp和53%1,2-乙烯基水平(基于bd63wt.%的总乙烯基含量)。当向配方中添加每摩尔活性锂0.2当量的叔戊醇钠(sta)时,vcp水平达到23%,同时将1,2乙烯基保持在53%(基于bd76%的总乙烯基含量)。因此,tg从含有低vcp的sbr中的-31℃升高至含有高vcp的sbr中的-14℃。中国专利10845109显示在环己烷溶液中,在正丁基锂(n-buli)和n,n-二甲基-四氢糠基胺(dmthfma)存在下,采用6/1的dmthfma/n-buli比率、大约7.7的溶剂与单体的比率和60℃的聚合温度使丁二烯分批聚合,产生85%的1,2-乙烯基富集率,但代价是反应时间为2小时且分子量分布朝向1.13的多分散性mw/mn指数加宽。将高乙烯基聚丁二烯基锂产物与过量四氯化硅偶合的尝试获得66wt%的最大偶合效率。在该专利中没有考虑vcp含量。该专利中也没有对极性改性剂体系制备共轭二烯单体和乙烯基芳族单体的嵌段共聚物的能力进行评价。美国专利号5336739显示了使用作为极性改性剂的乙基四氢糠基醚(ete)、正丁基锂引发剂和己烷与单体比率为4进行的高乙烯基聚丁二烯和聚异戊二烯的合成。对于异戊二烯均聚,所获得的3,4+1,2加成为70%,ete/li摩尔比为5且反应温度控制在70℃。在此类条件下聚合的聚丁二烯的1,2-乙烯基含量达到76%;当ete/li比率升高至10,同时将聚合温度降低至60℃时,获得了具有80%1,2-乙烯基富集的聚丁二烯。美国专利申请2012/0004379a1显示,当在正丁基锂(n-buli)存在下在相当高的温度下合成聚丁二烯和苯乙烯-丁二烯无规共聚物(sbr)时,基于双[2-(n,n-二甲基氨基)乙基]醚(bdmaee)和醇钠(如3,7-二甲基辛醇钠(na-dmo)或如薄荷醇钠(smt))的二元极性改性剂体系可用于获得非常高的1,2-乙烯基含量。显示了当采用3/0.5/1的bdmaee/na-dmo/n-buli摩尔比时乙烯基含量高达94wt%,以及当用smt替代制剂中的na-dmo时乙烯基含量为93wt%的聚丁二烯的实例。将该极性改性剂体系与高乙烯基聚丁二烯和无规苯乙烯-丁二烯共聚物中诸如以下的其它极性改性剂体系进行了比较测试:单独的bdmaee、单独的na-dmo、tmeda/na-dmo、tmeda/smt、dthfp/na-dmo,并且在乙烯基增强能力方面超过了所有其它极性改性剂体系。然而,还提到在80℃下聚合60分钟后的最终转化并不完全;在bdmaee/na-dmo/n-buli摩尔比为3/0.5/1下进行的丁二烯均聚仅达到97%的转化。另外,显示当以固定的n-buli剂量增加bdmaee/n-buli或na-dmo/n-buli的比率时,门尼粘度(mooneyviscosity)显著上升。单体完全转化的不足和门尼粘度对极性改性剂剂量的高依赖性两者都是这些极性改性剂引起阴离子活性中心中毒的信号。该专利中没有关于所获得的vcp水平的报道,也没有关于聚合物阴离子对于偶合剂的性能或证明有聚合活性的其它手段的性能的报道。该专利申请中没有提到该极性改性剂体系用于合成共轭二烯单体和乙烯基芳族单体的嵌段共聚物的有用性。kozak等(internationaljournalofpolymercharacterization,第20卷第7期,(2015),602-611和internationaljournalofpolymercharacterization,第21卷第1期,(2016),44-58)比较了以下各种极性改性剂体系在70℃下在15%丁二烯溶液聚合中的性能:dthfp、dthfp/smt、tmeda/smt、2,2-双(2,2-二甲基氨基乙基)醚(bdmaee)和bdmaee/smt,以及其它各种体系。发现bdmaee/smt是丁二烯锂引发聚合中1,2-乙烯基增强方面最强大的极性改性剂体系,达到约90%的乙烯基含量。单独的bdmaee具有差性能,达到约70%的乙烯基含量,但反应2h后转化较差。另一值得注意的发现是,当与单独的dthfp比较时,组合dthfp和smt对乙烯基含量水平具有不利作用。halasa和hsu(polymer,43,(2002),7111-7118)指出,当与tmeda组合时,任何种类的醇盐可同样有效地加速共轭二烯反应并获得高乙烯基含量。同样,在丁二烯的情况下,据报道,当在tmeda/li摩尔比大于或等于2和烃氧基化物(alcoxide)/li摩尔比大于或等于0.5的情况下,在75℃下使丁二烯聚合时,乙烯基水平为82-83。据报道,与使用基于乙基四氢糠基醚/smt(ete,具有两个醚部分的分子)的体系相比时,当组合smt与tmeda时,tg值更高,表明乙烯基含量更高,且丁二烯聚合速率更高。值得注意的是,在他们的研究中报告的乙烯基含量包括2%至6%的vcp。使用本发明生产的高乙烯基嵌段共聚物可应用于各种高端值领域,例如:如ep1730201和us7,439,301中所提到的用于高流动性sebs共聚物的前体。这种sebs特别可用于制造需要避免使用可能潜在地迁移并影响使用者的使用、触觉和/或健康的增塑剂和低分子量添加剂的制品。此外,sbs共聚物前体中的高乙烯基含量允许生产如专利ep1002813中所提到的展现出与聚丙烯的高相容性的sebs,使得它们非常适用于制造弹性薄膜、纤维和非织造物化合物;偶合的高乙烯基sbs共聚物可用作旨在用于软质弹性体薄膜的sebs的前体,如us22311767中所示;用于供电缆填充化合物中使用的油凝胶组合物中的sebs共聚物的前体,如ep0822227中所示;用于旨在用于密封剂制剂的sebs的前体,如us5,777,043中所示;当与聚烯烃复合时提供高透明度和改进的机械性质的sebs的前体,如us2010/0331465a1中所示;用于配制具有吸氧能力的聚丙烯组合物,如us2012/0252922a1中;用于辐射可固化热熔粘合剂组合物中,如us6,486,229中所示;用于热熔压敏粘合剂中,如us2015/0191637a1中。技术实现要素:已出人意料地发现,基于三元共混物(dthfp/bdmaee/smt、ete/bdmaee/smt、tmeda/bdmaee/smt和dmthfma/bdmaee/smt)的极性改性剂体系特别有利于制备共轭二烯单体和乙烯基芳族单体的嵌段共聚物。可获得共轭二烯单体的非常高水平的1,2-乙烯基和3,4-乙烯基键加成、低乙烯基环戊烷含量、单峰窄分子量分布和低水平的无规化重复单元掺入。此外,本发明使得能够以有竞争力的生产率且以低于现有技术的1,2-乙烯基和3,4-乙烯基加成度对温度的敏感性制备这种共聚物。尽管可在技术文献中发现bdmaee/smt体系可用于以快速聚合速率与可获得的乙烯基含量对聚合温度的低敏感性制备高乙烯基聚丁二烯,但我们的评价显示该极性改性剂不能制备乙烯基芳族单体和共轭二烯单体的嵌段共聚物,所述嵌段共聚物同时满足单峰窄分子量分布和无规化共聚单体低程度掺入以及竞争性聚合速度的要求。在制剂中存在该极性改性剂的临界浓度水平,超过该浓度水平,所述制剂产生分子量加宽和甚至双峰分子量分布共聚物的问题;当尝试使用高装料的bdmaee/smt生产三嵌段sbs共聚物时,产生低分子量二嵌段共聚物与高分子量三嵌段共聚物的共混物。低于该临界装料水平,bdmaee/smt能够生产具有窄分子量分布的嵌段共聚物,然而聚合速率变得相当慢;这具有产生具有相当高水平的无规化共聚单体掺入的嵌段共聚物的伴随缺点。本发明实现生产比用于生产高乙烯基含量sbs的当前路易斯碱(lewisbase)体系更纯的sbs三嵌段共聚物。与具有较不纯的嵌段的sbs或展现出较宽分子量分布的sbs相比,纯三嵌段sbs展现出突出的弹性性能。本发明实现制备具有高于常规方法的乙烯基含量的sebs。高乙烯基含量与相对低的分子量和中至低的苯乙烯含量的组合使sebs展现出流动性。高熔体流动性与高乙烯基含量的组合使得相比于在健康相关应用中相对于pvc有利地使用时,sebs与聚烯烃的共混物,特别是与聚丙烯的共混物更相容。此外,展现出高熔体流动性的sebs可作为纯聚合物或以化合物形式用于许多应用中,包括注射模制以生产医疗装置,用于个人护理的包覆模制应用,用于柔软触感材料、以及汽车零件、吹塑薄膜、浸渍的制品、以及薄膜和纤维。发现包含双四氢糠基丙烷(dthfp)与bdmaee和smt的极性改性剂体系展现出与现有技术体系相比的许多改进。dthfp是由下式(i)表示的许多化合物中的一种。尽管尚未完成实验工作,但有理由相信,由式(i)表示的许多其它化合物,如果不是所有的化合物,均可取代dthfp,预期会有类似的结果。其中r1至r14独立地为-h或-cnh2n+1基团,其中n=1至6。发现包含乙基四氢糠基醚(ete)与bdmaee和smt的极性改性剂体系展现出与现有技术体系相比的许多改进。ete是由下式(ii)表示的许多化合物中的一种。尽管尚未完成实验工作,但有理由相信,由式(ii)表示的许多其它化合物,如果不是所有的化合物,均可取代ete,预期会有类似的结果。其中r1是-cnh2n+1基团,其中n=1至6;并且其中r2至r7独立地为-h或-cnh2n+1基团,其中n=1至6。1.发现包含n,n,n’,n’-四甲基乙二胺(tmeda)与bdmaee和smt的极性改性剂体系展现出与现有技术体系相比的许多改进。tmeda是由下式(iii)表示的许多化合物中的一种。尽管尚未完成实验工作,但有理由相信,由式(iii)表示的许多其它化合物,如果不是所有的化合物,均可取代tmeda,预期会有类似的结果。其中r1至r4独立地为-ch3或-ch2ch3基团;并且其中r5是-h或-cnh2n+1基团,其中n=1至2。2.发现包含四氢糠基-n,n-二甲胺与bdmaee和smt的极性改性剂体系展现出与现有技术体系相比的许多改进。四氢糠基-n,n-二甲胺是由下式(iv)表示的许多化合物中的一种。尽管尚未完成实验工作,但有理由相信,由式(iv)表示的许多其它化合物,如果不是所有的化合物,均可取代四氢糠基-n,n-二甲胺,预期会有类似的结果。其中r1至r6独立地为-h或-cnh2n+1基团,其中n=1至6;并且其中r7和r8独立地为-cnh2n+1基团,其中n=1至2。3.发现包含dthfp、ete、tmeda和/或四氢糠基-n,n-二甲胺与bdmaee和smt的极性改性剂体系展现出与现有技术体系相比的许多改进。bdmaee是双[2-(n,n-二甲基氨基)乙基]醚,是由下式(v)表示的许多化合物中的一种。尽管尚未完成实验工作,但有理由相信,由式(v)表示的许多其它化合物,如果不是所有的化合物,均可取代bdmaee,预期会有类似的结果。其中m=1至2,n=1至2,并且其中r1至r4独立地为-cnh2n+1基团,其中n=1至6。具体实施方案以下描述本发明如何实现、如何起作用以及如何利用本发明。讨论了本发明的替代实施方式。如果本发明被认为可适用于化合物族,则确定该化合物族,并讨论可以预期实现同样的结果的原因。可通过本领域已知的任何适合的方法(如美国专利号3,281,383和3,753,936中所述的那些)来制备阴离子聚合的聚合物。在这些方法中,通过使阴离子可聚合的单体与作为引发剂的有机锂化合物接触来制备阴离子聚合的聚合物。这些化合物的优选类别可由式rli表示,其中r是选自由含有1至20个碳原子的脂族、脂环族和芳族基团组成的组的烃基,尽管可使用更高分子量的引发剂。许多阴离子聚合引发剂是众所周知的且可从市面购得。单官能有机锂化合物(如丁基锂)是常用引发剂的实例。这些引发剂的特定实例包括甲基锂、乙基锂、叔丁基锂、仲丁基锂、正丁基锂、正癸基锂、异丙基锂、二十烷基锂、环烷基锂化合物(如环己基锂)和芳基锂化合物(如苯基锂、萘基锂、对甲苯基锂、1,1-二苯基己基锂等)。被受保护极性官能团取代的单官能有机锂化合物也可用作用于阴离子聚合的引发剂。引发剂的量取决于阴离子聚合的聚合物的期望分子量而变化。可通过由因子100/(单体的mw)校正的每摩尔单体添加约0.20毫摩尔至5.0毫摩尔rli引发剂来获得约20,000至500,000的数均分子量。多官能有机锂引发剂也可用作制备具有期望的官能度范围的支化和星形共聚物的引发剂,所述官能度范围为每个引发剂分子2至约30条阴离子聚合的聚合物链。通过化学计量量的单官能有机锂化合物与多乙烯基化合物如1,3-二异丙烯基苯、1,3,5-三异丙烯基苯、1,3-双(1-苯基乙烯基)苯、1,3,5-三(1-苯基乙烯基)苯、1,3-二乙烯基苯、1,3,5-三乙烯基苯等的直接加成反应,可容易地制备多官能有机锂引发剂。低聚聚乙烯化合物可用于制备具有高官能度的多官能有机锂引发剂。单官能有机锂化合物(如丁基锂)是用于上述加成反应的常用引发剂的实例。这些常用引发剂的特定实例包括叔丁基锂、仲丁基锂和正丁基锂。被受保护极性官能团取代的单官能有机锂化合物也可用于制备多官能有机锂引发剂。多官能有机锂化合物可在它们之间和/或与单官能有机锂化合物组合,以部分地引发与多官能有机锂化合物的阴离子聚合。部分引发是通过控制多官能引发剂与单官能引发剂的化学计量比来实现的。阴离子聚合通常在惰性烃溶剂中,在相对低的温度下,在真空或惰性气氛下,用高度纯化的试剂进行,以防止聚合反应过早终止。阴离子聚合反应可在多种有机溶剂中进行。适合溶剂的实例包括但不限于戊烷、己烷、庚烷、辛烷、环戊烷、环己烷、环庚烷、苯、萘、甲苯、二甲苯、甲醚、甲基乙基醚、乙醚、四氢呋喃、丙酮、甲基乙基酮和其混合物。环己烷特别适合用作阴离子聚合中的溶剂。阴离子聚合通常在-100℃至150℃、优选-75℃至75℃范围内的温度下进行。通常使用50重量%至90重量%的反应溶剂来控制反应区内的粘度,优选70%至85%。阴离子聚合的典型停留时间取决于反应温度和引发剂水平在0.1小时与5小时之间、优选从0.2小时至2小时变化。用于构建本发明的阴离子聚合的聚合物的适合共轭二烯包括但不限于1,3-丁二烯、异戊二烯、1,3-戊二烯、甲基戊二烯、苯基丁二烯、2,3-二甲基-1,3-丁二烯、2,4-己二烯、1,3-己二烯、1,3-环己二烯、3,4-二甲基-1,3-己二烯、1,3-辛二烯、4,5-二乙基-1,3-辛二烯、香叶烯、金合欢烯(farnesene)等。可用于生产阴离子聚合的聚合物的其它阴离子可聚合的单体包括但不限于单乙烯基芳族单体,如苯乙烯和苯乙烯衍生物,包括3-甲基苯乙烯、α-甲基苯乙烯、对甲基苯乙烯、α,4-二甲基苯乙烯、叔丁基苯乙烯、邻氯苯乙烯、2-丁烯基萘、4-叔丁氧基苯乙烯、3-异丙烯基联苯、4-乙烯基吡啶、2-乙烯基吡啶和异丙烯基萘、4-正丙基苯乙烯。可用于生产阴离子聚合的聚合物的官能化共轭二烯单体和官能化单乙烯基芳族单体包括但不限于甲硅烷基化单体等。在这里提供的方法的一些实施方案中,阴离子聚合的聚合物经历全部或部分偶合以制备支化和星形阴离子聚合的聚合物。部分偶合意指全部活性阴离子聚合的聚合物链端的一部分经历与偶合剂偶合。偶合剂期望地在2条和30条阴离子聚合的聚合物链之间偶合,尽管还可采用能够使更大数目的链偶合的偶合剂。用于全部或部分偶合步骤中的适合偶合剂包括但不限于环氧化大豆油、二乙烯基苯、卤化锡、卤化硅、官能化锡化合物、官能化硅化合物(如硅烷化合物)和官能化低聚化合物(如美国专利号7,517,934中所列的那些)。美国专利号7,517,934的全部公开内容以引入方式并入本文中。四氯化硅和四氯化锡是适合偶合剂的特定实例,其中四氯化硅特别适合于该应用。通过控制偶合剂与活性聚合物的化学计量比实现部分偶合。部分偶合可提供具有期望性质的聚合物共混物。当与烷基锂引发剂混合时,可使用来自第iia族、第iib族和第iiia族的不同金属(包括镁、锌和铝)的有机金属化合物作为聚合速率调节剂。适合的聚合速率调节剂的特定实例是二丁基镁、二乙基锌和三乙基铝。聚合速率调节剂可用于控制聚合的温度曲线。聚合速率调节剂有助于以预确立停留时间的等温模式或直至峰值温度的准绝热模式控制聚合步骤。在这里提供的方法的一些实施方案中,阴离子聚合的聚合物以分批、程序化分批和/或半分批工艺聚合。在本发明方法的另外的实施方案中,可以连续和/或半连续模式制备阴离子聚合的聚合物。阴离子聚合的聚合物的阴离子聚合可原位发生,即在单个反应区中发生,或者可在多个反应区中发生。前一种设计倾向于有利于更快的反应,而当期望特别控制的聚合反应时,后一种设计可能是优选的。在一些实施方案中,可使用具有两个或更多个反应区(例如,反应室)的反应设备。本领域技术人员将认识到,所描述的阴离子聚合的聚合物的合成可在反应设置中进行,该反应设置包括在达到所述停留时间和化学计量条件所需的温度、溶剂比和物流流速下操作的分批、半连续或连续工艺。以下实施例具有显示本发明特征的目的,且并不意在限制其范围。使用现有技术的比较实施例被包括作为参考。通过300mhz1h-nmr技术进行共聚物微观结构的表征,并且使用具有与差示折射率检测器耦合的3柱组的gpc进行分子量表征。所报告的峰值分子量和多分散性指数mw/mn参考基于具有窄分子量分布的聚苯乙烯标准品的校准曲线。实施例1:使用本发明优选的极性改性剂体系的未偶合的sbs嵌段共聚物:低温范围内的高极性改性剂体系浓度的高分子量共聚物合成。在惰性氮气气氛下,将环己烷(5383g)装载至7.6升不锈钢反应器中。借助于通过反应器内部盘管的水循环,使溶剂温度在反应器中在18.1℃(tst1)下稳定。此后,以所列出的顺序添加bdmaee、dthfp、正丁基锂引发剂(nbuli)引发剂和smt。dthfp、bdmaee和smt相对于活性锂含量的摩尔比分别为4.17、0.52和0.10。将苯乙烯(84.5g)以足以在0.5分钟内完成其装料的速率进给至反应器中。沿着第一苯乙烯嵌段聚合继续在受控温度下循环水。反应器温度在3分钟内达到19.7℃的峰值温度,并且实施2分钟的等待时间,因此第一嵌段均聚时间为5min(tst)。然后在约2分钟的时期内进给丁二烯(344.7g)后立即终止反应器冷却。丁二烯进料开始时的温度为19.2℃(ti-bd)。丁二烯聚合在开始丁二烯装料后9分钟(tp-bd)达到43℃的峰值温度(tp-bd)。在每种情况下,丁二烯聚合均在没有水循环通过夹套的情况下发生。在将第二苯乙烯(84.5g)进给至反应器之前,有3分钟的等待时间(twbd)。在3分钟的第二苯乙烯装料后检测到峰值温度。在5分钟之后,进给醇以终止聚合物阴离子。该程序中环己烷与总单体的质量比为8。获得了具有以下特征的嵌段共聚物:峰值分子量mp=277.8kg/mol,多分散性指数mw/mn=1.06,总苯乙烯重复单元含量为31.9wt%,无规苯乙烯重复单元含量为3.9wt%,基于丁二烯嵌段的总乙烯基含量为90.9wt%(1,2-乙烯基+vcp),以及基于重复单元的总乙烯基含量为89.4mol%,vcp含量为3.1wt%。该sbs共聚物的分子量分布展现单峰窄峰形状。实施例2:使用本发明优选的极性改性剂体系的未偶合的sbs嵌段共聚:低温范围内的中极性改性剂浓度范围的中分子量共聚物合成。以5.3升实验室规模批料通过以下步骤制备嵌段共聚物:(1)将环己烷溶剂装载至反应器中;(2)将溶剂稳定至温度ti;(3)装载极性改性剂bdmaee和dthfp;(4)装载正丁基锂;(5)装载smt;(6)装载苯乙烯单体;(7)立即中断反应器温度控制,以除了自然热量损失至环境中之外,在不存在外部冷却或外部加热的情况下进行聚合;(8)将第一苯乙烯嵌段均聚时段tst1,其中在此期间聚合热逐渐升高反应器温度,而没有检测到证明峰值温度的温度下降;(9)记录反应器温度tibd并立即装载丁二烯单体;(10)丁二烯嵌段共聚,其中反应热将反应器温度升高至峰值温度tpbd,记录该峰值温度tpbd以及从丁二烯装料开始至出现tpbd的时刻所经过的时间tpbd;(11)等待时间twbd;(12)装载第二苯乙烯装料;(13)使第二聚苯乙烯嵌段聚合,直至苯乙烯单体完全消耗;(14)装载过量当量的醇以确保终止所有聚合物阴离子。在该制剂中,使用2938g环己烷,将60.9g苯乙烯用于构建聚苯乙烯第一嵌段,将246.1丁二烯用于构建高乙烯基聚丁二烯中间嵌段,并且将60.9g苯乙烯用于构建聚苯乙烯末端嵌段。因此,总溶剂与单体的比率(s/m)为8.0。每次苯乙烯装料的进料时间长0.5分钟。丁二烯装料的进料时间长2分钟。活性正丁基锂载量是4.2mmol。dthfp、bdmaee和smt相对于活性锂含量的摩尔比分别为2.13、0.27和0.05(参见表1)。首先使苯乙烯装料从10.8℃起始温度均聚3分钟,达到15.0℃。高乙烯基聚丁二烯嵌段聚合在11分钟内从15℃的温度发展至46.7℃的峰值温度(参见表2)。获得了具有以下特征的嵌段共聚物:总苯乙烯重复单元含量为34.6wt%,无规苯乙烯重复单元含量为3.3wt%,基于丁二烯嵌段的总乙烯基含量为87.6wt%(1,2-乙烯基+vcp),基于重复单元的总乙烯基含量为85.8mol%,基于丁二烯嵌段的vcp含量为3.7wt%。(参见表3)该sbs共聚物的分子量分布展现单峰窄峰形状,分子量mp=142.3kg/mol,且多分散性指数mw/mn=1.03;(参见表4)。比较实施例c1:在低温范围内使用最接近的现有技术极性改性剂体系bdmaee/smt的未偶合sbs嵌段共聚。在实施例2的相同的5.3升反应器中制备嵌段共聚物。采用实施例2的相同程序,以及相同量的环己烷、丁二烯和苯乙烯装料。评价现有技术的极性改性剂体系bdmaee/smt。如实施例2中那样,在正丁基锂引发剂之前进给bdmaee,并且在正丁基锂之后进给smt。活性正丁基锂载量是4.4mmol。bdmaee和smt相对于活性锂含量的摩尔比分别为0.59和0.05(参见表1)。第一苯乙烯装料均聚从10.7℃的起始温度达到15.0℃需要11分钟。高乙烯基聚丁二烯嵌段聚合在17分钟内从15℃的温度发展至46.0℃的峰值温度(参见表2)。获得了具有以下特征的嵌段共聚物:总苯乙烯重复单元含量为33.7wt%,无规苯乙烯重复单元含量为4.3wt%,基于丁二烯嵌段的总乙烯基含量为90.6wt%(1,2-乙烯基+vcp),基于重复单元的总乙烯基含量为88.7mol%,基于丁二烯嵌段的vcp含量为3.8wt%。(参见表3)该sbs共聚物的分子量分布展现双峰形状,其主峰在分子量mp=123.7kg/mol处,且次峰在mp2=178.9kg/mol处,且多分散性指数mw/mn=1.08;(参见表4和图2)。在该比较实施例中获得的sbs的总乙烯基含量几乎达到本发明实施例1中获得的90.9wt%水平。无规苯乙烯含量与总苯乙烯含量的比率与实施例1中获得的比率也非常相似:在比较实施例中,使用bdmaee/smt体系,约12.8%苯乙烯被丁二烯无规化,且87.2%苯乙烯成为聚苯乙烯嵌段的一部分,而在本发明实施例1中,12.2%苯乙烯为无规的且87.8%苯乙烯呈嵌段形式。然而,这在比较产物中是以不可接受的属性为代价的:比较实施例1中的双峰和宽分子量分布是聚合物阴离子过早终止的信号;因此,所生产的共聚物很可能由过早停止延伸的低分子量二嵌段共聚物sb和具有不对称大小的聚苯乙烯嵌段的高分子量sbs的共混物组成。这两个因素都会损害共聚物的机械性质。利用该比较极性改性剂体系时,聚合也变得不利地缓慢:在比较实施例1中使第一苯乙烯嵌段聚合需要15分钟,而在本发明实施例2中需要3分钟;利用bdmaee/smt体系时,丁二烯聚合也较慢,在比较实施例中达到峰值温度需要17分钟,而对于相同的温度升高,实施例2中的本发明体系需要11分钟。比较实施例c2:在低温范围内使用最接近的现有技术极性改性剂体系bdmaee/smt的未偶合sbs嵌段共聚。如实施例2中那样,在正丁基锂引发剂之前进给bdmaee,并且在正丁基锂之后进给smt。活性正丁基锂载量是4.4mmol。bdmaee和smt相对于活性锂含量的摩尔比分别为0.36和0.05(参见表1)。在15分钟的第一苯乙烯嵌段均聚后,反应器温度几乎没有从10.5℃的起始温度达到13.6℃。高乙烯基聚丁二烯嵌段聚合在42分钟内从13.6℃的温度发展至41.2℃的峰值温度(参见表2)。获得了具有以下特征的嵌段共聚物:总苯乙烯重复单元含量为34.9wt%,无规苯乙烯重复单元含量为10.6wt%,基于丁二烯嵌段的总乙烯基含量为87.0wt%(1,2-乙烯基+vcp),基于重复单元的总乙烯基含量为85.0mol%,基于丁二烯的vcp含量为4.2wt%。(参见表3)该sbs共聚物的分子量分布展现略微偏向低分子量范围的单峰型峰,其主峰在分子量mp=154.5kg/mol处,且多分散性指数mw/mn=1.05;(参见表4和图2)。比较实施例c2相对于比较实施例c1的主要变化是bdmaee与活性nbuli的摩尔比从0.59降低至0.36,而smt/nbuli摩尔比保持在0.05。这将乙烯基含量从比较实施例c1中的90.6降低至比较实施例c2中的87.0。降低乙烯基含量的作用与将本发明的三元极性改性剂体系的剂量从发明实施例1中4.17/0.52/0.1/1的dthfp/bdmaee/smt/nbuli摩尔比以一半降低至发明实施例2中的2.13/0.27/0.05/1的作用非常相似,分别有90.9wt%总乙烯基至87.6wt%的相关下降。比较实施例c2中相对于比较实施例c1中的分子量分布得到改进,因为避免了双峰性,但分布中仍然存在的一定偏斜证明了一些过早的聚合物阴离子终止。如在图2中可以看出,本发明实施例1中用极性改性剂dthfp/bdmaee/smt获得的sbs的分子量分布比在比较实施例c2中生产的共聚物的分子量分布更窄且更对称。比较实施例c2配方中bdmaee含量轻微降低的最有害作用是在聚合速度和无规化苯乙烯含量方面:比较实施例c2中允许第一苯乙烯嵌段均聚的时间与本发明实施例2中所需的时间相比增加了5倍,并且甚至这也不足以达到预计的15℃目标。比较实施例c2中的丁二烯聚合也非常慢,达到丁二烯峰值温度需要42分钟,而在本发明实施例2中仅需要11分钟。尽管在进给第二苯乙烯装料之前,在比较实施例c2中允许比本发明实施例2中更长的丁二烯峰值温度后的等待时间,分别为5分钟和2分钟,但无规化苯乙烯非常高:30.4%的苯乙烯被无规化,仅69.6%成为聚苯乙烯嵌段的一部分,而在本发明实施例2中,仅9.5%的苯乙烯变得无规化(计算为相对于基于sbs的总苯乙烯含量的基于sbs的无规苯乙烯含量)。在实施例2的相同的5.3升反应器中制备嵌段共聚物。采用实施例2的相同程序,以及相同量的环己烷、丁二烯和苯乙烯装料。以低于比较实施例c1中的剂量评价现有技术的极性改性剂体系bdmaee/smt。表1.根据本发明的实施例1和2以及使用现有技术的比较实施例c1至c6中的引发剂和极性改性剂体系装料表2.根据本发明的实施例1和2以及使用现有技术的比较实施例c1至c6中的聚合工艺参数表3.根据本发明的实施例1和2以及使用现有技术的比较实施例c1至c6中产生的sbs共聚物的nmr表征表4.根据本发明的实施例1以及使用现有技术的比较实施例c1至c6中产生的sbs共聚物的gpc表征实施例mp,kg/molmw/mnmwd形状1277.41.06单峰窄峰2142.31.03单峰窄峰c1123.71.08双峰型峰,mp2=179kg/molc2154.51.05单峰型峰,略微偏向低mwc3138.51.03单峰窄峰c4123.71.02单峰窄峰c5150.61.04单峰窄峰c6130.31.04单峰窄峰图2.根据本发明的实施例2以及使用现有技术的比较实施例c1和c2中产生的共聚物的分子量分布。当相对于比较实施例c1中使用的基于bdmaee/smt的现有技术的现有技术改性剂体系,比较用实施例2中使用的本发明的极性改性剂体系dthfp/bdmaee/smt获得的高乙烯基共聚物的苯乙烯均聚阶段的速度时,可注意到,本发明在几乎比现有技术的时段短4倍的时段内发生第一苯乙烯嵌段均聚的温度上升至15℃。当比较达到丁二烯嵌段阶段峰值温度的时间tpbd时,实施例2中测试的本发明的极性改性剂使得也能够比用于实施例c1中的现有技术更快地进行丁二烯聚合。另外,实施例2中的本发明极性改性剂需要比实施例c1中实施的现有技术更短的等待时间twbd,以获得sbs共聚物中更低的无规苯乙烯含量。此外,在实施例1中,bdmaee/li比率是比较实施例c1中所用比率的一半。当相对于比较实施例c1的基于极性改性剂体系bdmaee/smt的现有技术,比较用于实施例2中使用的本发明极性改性剂dthfp/bdmaee/smt获得的分子量分布时,可注意到,由本发明获得单峰窄峰,而由现有技术获得宽双峰型峰(参见图2)。在实施例2中用本发明的极性改性剂获得的单峰窄分布分子量分布确认,如此生产的多数sbs共聚物分子生长至相同的平均分子量,并且很可能呈对称的s-b-b-b-s共聚物。通过聚合物阴离子沿聚合过早终止来解释由现有技术获得的双峰和宽分子量分布:落在双峰分布的较低分子量范围内的共聚物链易于缺少聚苯乙烯末端嵌段的掺入,而那些落在所述分布的高分子量范围内的共聚物链易于具有比它们首先被掺入的聚苯乙烯嵌段更长的聚苯乙烯末端嵌段。然后,预计比较实施例c1的产物是具有大于50wt%的s-b-b二嵌段共聚物和小于50wt%的不对称s-b-b-b-s三嵌段共聚物的共混物。在评价现有技术的bdmaee/smt极性改性剂体系(其bdmaee/li比率低于比较实施例c1、但bdmaee的量仍高于本发明实施例2)的比较实施例c2中,聚合以更低的速度进行并且在共聚物中掺入多得多的无规苯乙烯重复单元。其分子量分布朝向单峰型峰改进,但仍然看起来比本发明实施例2的分子量分布稍宽,并偏向于低分子量范围,证明聚合物阴离子有些过早终止。基于dthfp/bdamee/smt极性改性剂体系的本发明实施例2产生具有非常高水平的1,2-乙烯基丁二烯重复单元的sbs嵌段共聚物,略高于基于现有技术的极性改性剂体系bdmaee/smt的比较实施例c2中所获得的1,2-乙烯基丁二烯重复单元。有利地,本发明实施例2中的vcp水平略低于比较实施例c1和c2中的vcp水平。当相对于其它现有技术的极性改性剂体系,如比较实施例c3中的dthfp、比较实施例c4中的ete、比较实施例c5中的tmeda/smt和比较实施例c6中的dmthfma/smt,比较基于dthfp/bdmaee/smt极性改性剂体系的本发明实施例2时,可注意到,本发明的极性改性剂体系比多数现有技术的极性改性剂体系进行更快的聚合,并且仅比dmthfma/smt体系略慢。通过本发明获得的共聚物的分子量分布单峰性和窄性与这些其它极性改性剂体系是有竞争优势的,如通过多分散性指数mw/mn的低值所揭示的。然而,这些其它现有技术的极性改性剂体系都不能超过本发明的1,2-乙烯基掺入水平。此外,在比较实施例c5中尝试的tmeda/smt和dmthfma/smt产生高于本发明实施例1的水平的vcp。实施例3至8:在中温范围内使用本发明的优选极性改性剂体系的未偶合sbs嵌段共聚。在实验室规模批料反应器中通过以下步骤制备s-b-b-b-s型嵌段共聚物:(1)将环己烷溶剂装载至反应器中;(2)将溶剂稳定至温度ti;(3)装载极性改性剂bdmaee和dthfp;(4)装载正丁基锂;(5)装载smt;(6)装载苯乙烯单体;(7)立即中断反应器温度控制,以除了自然热量损失至环境中之外,在不存在外部冷却或加热的情况下进行聚合;(8)第一苯乙烯嵌段的均聚,其中聚合热将反应器温度在从苯乙烯装料开始至峰值温度tpst所经过的时间tpst内逐渐升高直至该峰值温度;(9)tpst之后10分钟的等待时间,其中出现微小的温度下降;(10)记录反应器温度tibd并立即装载丁二烯单体;(11)丁二烯嵌段共聚,其中反应热将反应器温度升高至峰值温度tpbd,记录该峰值温度tpbd以及从丁二烯装料开始至出现tpbd的时刻所经过的时间tpbd;(12)tpbd之后1分钟的等待时间,其中出现微小的温度下降;(13)装载第二苯乙烯装料;(14)使第二聚苯乙烯嵌段聚合,直至苯乙烯单体完全消耗;(15)装载过量当量的醇以确保终止所有聚合物阴离子。在该制剂中,使用2795g环己烷,将63.6g苯乙烯用于构建聚苯乙烯第一嵌段,将246.1丁二烯用于构建高乙烯基聚丁二烯中间嵌段,并且将63.6g苯乙烯用于构建聚苯乙烯末端嵌段。因此,总溶剂与单体的比率为7.5。每次苯乙烯装料的进料时间长0.5分钟。丁二烯装料的进料时间长2分钟。活性引发剂含量以及极性改性剂与引发剂的摩尔比示于表5中。使高乙烯基聚丁二烯嵌段在22.8至56.8℃的温度范围内聚合。工艺参数(ti、tpst、tpst、tibd、tpbd和tpbd)列于表6中。来自该实施例的产物的sbs共聚物表征示于表7和表8中。实施例9至11:在中温范围内使用本发明的替代极性改性剂体系的未偶合sbs嵌段共聚。采用实施例3至7中所列出的相同操作,但dthfp被ete、tmeda或dmthfma替代。用于这些实施例的活性正丁基锂和极性改性剂体系剂量报告于表5中。使高乙烯基聚丁二烯嵌段在23至59.8℃的温度范围内聚合。每个实施例的工艺参数(ti、tpst、tpst、tibd、tpbd和tpbd)列于表6中。来自这些实施例的产物的sbs共聚物表征示于表7和表8中。比较实施例c7和c8.使用最接近的现有技术的极性改性剂体系,在中温范围内进行未偶合的sbs嵌段共聚合。采用实施例3至7中所述的相同程序,但避免dthfp。用于这些比较实施例的活性正丁基锂和极性改性剂体系剂量报告于表5中。使高乙烯基聚丁二烯嵌段在23.5至57℃的温度范围内聚合。每个比较实施例的工艺参数(ti、tpst、tpst、tibd、tpbd和tpbd)列于表6中。来自这些比较实施例的产物的sbs共聚物表征示于表7和表8中。表5.根据本发明的实施例3至11以及使用现有技术的比较实施例c7和c8中的引发剂和极性改性剂体系装料表6.根据本发明的实施例3至11以及使用现有技术的比较实施例c7和c8中的聚合工艺参数表7.根据本发明的实施例3至11以及使用现有技术的比较实施例c7和c8中制备的sbs共聚物的nmr表征表8.根据本发明的实施例3至11以及使用现有技术的比较实施例c7和c8中制备的sbs共聚物的gpc表征图3.根据本发明的实施例3以用使用现有技术的比较实施例c7和c8中生产的共聚物的分子量分布。在本发明实施例3至8中,基于极性改性剂体系dthfp/bdmaee/smt,dthfp、bdmaee和smt相对于活性锂的极性改性剂比率在宽的组成范围内变化。当相对于使用现有技术的bdmaee/smt极性改性剂体系的比较实施例c7和c8,比较基于dthfp/bdmaee/smt的本发明实施例3至8的聚合速度时,用本发明极性改性剂体系尝试的多数组合物达到第一苯乙烯嵌段峰值温度的聚合时间更短,并且达到丁二烯聚合峰值温度的时间更短。有利地,基于dthfp/bdmaee/smt极性改性剂体系的本发明实施例3至8的分子量分布在每种情况下都展现出具有低多分散性指数mw/mn的窄单峰型峰形,而基于现有技术bdmaee/smt极性改性剂体系的比较组合物c7和c8产生具有高多分散性指数mw/mn的宽双峰分子量分布。图3显示了该行为的相关比较:本发明实施例3和比较实施例c8具有相同的活性锂、bdmaee和smt含量;在本发明实施例3中掺入dthfp校正了比较实施例c8的宽双峰分子量分布。比较实施例c7中较高剂量的bdmaee使分子量分布加宽略微恶化,如通过图3和多分散性指数mw/mn所揭示的。因此,现有技术不利地易于产生比本发明组合物更高含量的不期望的二嵌段s-b-b共聚物和非对称s-b-b-b-s三嵌段共聚物。本发明实施例9至11显示可替代dthfp/bdmaee/smt极性改性剂体系的dthfp组分的适当极性改性剂的选择。即体系ete/bdmaee/smt、tmeda/bdmaee/smt和dmthfma/bdmaee/smt与dthfp/bdmaee/smt表现一样好,赋予非常高乙烯基含量的嵌段共聚物的高速聚合以及单峰窄分子量分布。dthfp/bdmaee/smt极性改性剂体系是优选的选择,因为ete/bdmaee/smt以略高的多分散性指数mw/mn进行,tmeda/bdmaee/smt在苯乙烯均聚步骤中滞后并产生较高的vcp含量,而dmthfma/bdmaee/smt产生略低的1,2-乙烯基含量。然而,该替代性本发明体系仍然超过所述现有技术系统的性能。尽管在本发明实施例3至11中比在比较实施例c2至c6中更高的温度下进行嵌段共聚,但与利用现有技术相比,利用本发明获得了1,2-乙烯基含量的更高掺入水平,还能够产生窄分子量分布的sbs共聚物。实施例12.在高温范围内使用本发明的优选极性改性剂体系的未偶合sbs嵌段共聚。在具有相对低的热绝缘水平的实验室规模的搅拌分批反应器中,使用以下程序制备s-b-b-b-s型嵌段共聚物:在氮气气氛下将环己烷(2720g)装载至搅拌反应器中,并将其温度稳定在43.3℃(ti)。此后,以所列出的顺序将bdmaee、dthfp、正丁基锂引发剂和smt添加至所述反应器中。活性引发剂含量以及极性改性剂与引发剂的摩尔比示于表9中。然后,将第一部分苯乙烯单体(56.4g)装载至反应器中。在0.5分钟内完成苯乙烯进料操作。此后停止反应器温度控制,因此除了向环境散热之外,在既不存在外部冷却也不存在外部加热的情况下进行聚合。在2分钟(tpst1)的苯乙烯装料后检测到46.2℃的峰值温度(tpst1)。然后,实施3分钟的等待时间。到那时,反应器温度下降至44.2℃(tibd)并开始丁二烯(227.5g)进料操作。在2分钟内完成丁二烯装载。从丁二烯进料操作开始计算,丁二烯聚合热在5分钟的时期(tpbd)内将反应物质量升高至61.1℃的峰值温度(tpbd)。然后,在丁二烯聚合峰值温度和第二苯乙烯装料开始之间有2分钟的等待时间(twbd)。完成至反应器中的第二苯乙烯装料(56.4g)需要30秒。使第二苯乙烯装料聚合足够长的时间以被完全消耗,然后将醇进给至反应器中以终止阴离子聚合物阴离子。在该实施例中,环己烷溶剂与总单体进料的比率(s/m)为8。工艺参数(ti、tpst1、tpst1、tibd、tpbd、tpbd和twbd)列于表10中。来自该实施例的产物的sbs共聚物表征示于表11和表12中。比较实施例c9.使用现有技术的极性改性剂体系,在高温范围内进行未偶合的sbs嵌段共聚合。在具有相对低的热绝缘水平的实验室规模的搅拌分批反应器中,使用以下程序制备s-b-b-b-s型嵌段共聚物:在氮气气氛下将环己烷(2862g)和苯乙烯单体(63.0g)装载至搅拌反应器中,并将其温度稳定在39.9℃的温度(ti)。此后,以所列出的顺序将tmeda、smt和正丁基锂引发剂添加至反应器中,并关闭反应器温度控制。活性引发剂含量以及极性改性剂与引发剂的摩尔比示于表9中。在5分钟(tpst1)的引发剂装料后检测到44.5℃的峰值温度(tpst1)。然后将反应器冷却15分钟以达到28℃的温度(tibd)。从此刻开始,聚合继续在除了向环境散热之外不存在预期加热或冷却的情况下进行。然后开始丁二烯(255.4g)进料操作。在5分钟内完成丁二烯装载。从丁二烯进料操作开始计算,丁二烯聚合热在10分钟的时期(tpbd)内将反应物质量升高至62.7℃的峰值温度(tpbd)。然后,在丁二烯聚合峰值温度和第二苯乙烯装料开始之间有5分钟的等待时间(twbd)。使至反应器中的第二苯乙烯装料(63.0g)聚合足够长的时间以被完全消耗。然后将醇进给至反应器中以终止阴离子聚合物阴离子。在该实施例中,环己烷溶剂与总单体进料的比率(s/m)为7.5。工艺参数(ti、tpst1、tpst1、tibd、tpbd、tpbd和twbd)列于表10中。来自该实施例的产物的sbs共聚物表征示于表11和表12中。表9.根据本发明的实施例12中以及使用现有技术的比较实施例c9中的引发剂和极性改性剂体系的装料,以及总溶剂与单体(s/m)的比率表10.根据本发明的实施例12和使用现有技术的比较实施例c9中的聚合工艺参数。表11.根据本发明的实施例12和使用现有技术的比较实施例c9中产生的sbs共聚物的nmr表征。表12.根据本发明的实施例12和使用现有技术的比较实施例c9中产生的sbs共聚物的gpc表征。基于极性改性剂体系dthfp/bdmaee/smt的实施例12与基于现有技术的极性改性剂体系tmeda/smt的比较实施例c9的比较显示,当在高聚合温度下进行时,本发明极性改性剂体系比现有技术以在聚丁二烯嵌段中更高1,2-乙烯基和低得多的vcp重复单元的掺入进行。此外,丁二烯聚合在实施例10中跨越44至61℃的温度范围,这是比比较实施例c9中跨越在28至62℃范围内的丁二烯聚合更苛刻的条件(更高的平均聚合温度)。两种情况(本发明和现有技术)均以单峰窄分子量分布进行。比较实施例c10.使用现有技术的极性改性剂体系,在高浓度下,在高温范围内进行未偶合的sbs嵌段共聚合。表13.使用现有技术的比较实施例c10中的引发剂和极性改性剂体系的装料,以及总溶剂与单体(s/m)的比率。表14.使用现有技术的比较实施例c10中产生的sbs共聚物的nmr表征。表15.使用现有技术的比较实施例c10中产生的sbs共聚物的gpc表征。实施例13和14.使用本发明的优选极性改性剂体系,在低单体浓度水平下进行未偶合的sbs嵌段共聚。在实验室规模批料反应器中通过以下步骤制备s-b-b-b-s型嵌段共聚物:(1)将环己烷溶剂装载至反应器中;(2)将溶剂稳定至温度ti;(3)装载极性改性剂bdmaee和dthfp;(4)装载正丁基锂;(5)装载smt;(6)装载苯乙烯单体;(7)立即中断反应器温度控制,以除了自然热量损失至环境中之外,在不存在外部冷却或加热的情况下进行聚合;(8)第一苯乙烯嵌段持续总时间tst的均聚,在此时段期间,聚合热将反应器温度以从苯乙烯装料开始至峰值温度tpst所经过的时间tpst逐渐升高直至该峰值温度;(10)记录反应器温度tibd并立即装载丁二烯单体;(11)丁二烯嵌段共聚,其中反应热将反应器温度升高至峰值温度tpbd,记录该峰值温度tpbd以及从丁二烯装料开始至出现tpbd的时刻所经过的时间tpbd;(12)tpbd之后twbd的等待时间,其中出现微小的温度下降;(13)装载第二苯乙烯装料;(14)使第二聚苯乙烯嵌段聚合,直至苯乙烯单体完全消耗;(15)装载过量当量的醇以确保终止所有聚合物阴离子。在这些制剂中,使用2805g环己烷,将41.8g苯乙烯用于构建聚苯乙烯第一嵌段,将171.1丁二烯用于构建高乙烯基聚丁二烯中间嵌段,并且将41.8g苯乙烯用于构建聚苯乙烯末端嵌段。因此,总溶剂与单体的比率为11.0。每次苯乙烯装料的进料时间长0.5分钟。丁二烯装料的进料时间长2分钟。活性引发剂含量以及极性改性剂与引发剂的摩尔比示于表16中。工艺参数(ti、tpst、tpst、tsttibd、tpbd、tpbd和twbd)列于表17中。来自该实施例的产物的sbs共聚物表征示于表18和表19中。比较实施例c11和c12.使用现有技术的极性改性剂体系,在低单体浓度水平下进行未偶合的sbs嵌段共聚合:采用了实施例10和11中所述的相同步骤,但避免使用dthfp。在这些制剂中,使用4228g环己烷,将68.2g苯乙烯用于构建聚苯乙烯第一嵌段,将287.7g丁二烯用于构建高乙烯基聚丁二烯中间嵌段,并且将68.2g苯乙烯用于构建聚苯乙烯末端嵌段。因此,总溶剂与单体的比率为10.0。用于这些比较实施例的活性正丁基锂和极性改性剂体系剂量报告于表16中。每个比较实施例的工艺参数(ti、tpst、tpst、tst、tibd、tpbd和tpbd)列于表17中。来自这些比较实施例的产物的sbs共聚物表征示于表18和表19中。表16.根据本发明的实施例13和14中以及使用现有技术的比较实施例c11和c12中的引发剂和极性改性剂体系的装料,以及总溶剂与单体(s/m)的比率。表17.根据本发明的实施例13和14以及使用现有技术的比较实施例c11和c12中的聚合工艺参数。n.d.=未检测到表18.根据本发明的实施例13和14以及使用现有技术的比较实施例c11和c12中产生的sbs共聚物的nmr表征。表19.根据本发明的实施例13和14以及使用现有技术的比较实施例c11和c12中产生的gpc共聚物的nmr表征。图4.根据本发明的实施例13和14以及使用现有技术的比较实施例c11和c12中产生的共聚物的分子量分布。基于本发明极性改性剂体系dthfp/bdmaee/smt的实施例13和14的分析显示它们在聚合期间对较低单体浓度稳健,以苯乙烯和丁二烯嵌段的高聚合速率产生聚丁二烯嵌段中的高1,2-乙烯基和低vcp重复单元含量,且最终产物中的单峰分子量分布窄。另一方面,采用现有技术的极性改性剂体系bdmaee/smt,在稀释单体状态下进行聚合,当bdmaee/li比率高时,聚合以双峰重量分布进行,或者当bdmaee/li低时,聚合以非常慢的速率以及在共聚物中非常高水平的无规苯乙烯掺入进行。实施例15和16.使用优选的极性改性剂体系进行sb嵌段共聚,之后进行偶合步骤。在实验室规模的搅拌反应器中,使用以下程序制备(s-b-b)n-x型共聚物:在氮气气氛下将环己烷(2792g)装载至搅拌反应器中,并将其温度稳定在大约18℃(ti)。此后,以所列出的顺序将bdmaee、dthfp、正丁基锂引发剂和smt添加至所述反应器中。活性引发剂含量以及极性改性剂与引发剂的摩尔比示于表20中。然后,将苯乙烯单体(63.6g)装载至反应器中。在30秒内完成苯乙烯进料操作。此后中断反应器冷却。记录第一苯乙烯均聚阶段的峰值温度(tpst)以及从第一苯乙烯装料开始直至该时刻所经过的反应时间(tpst)。然后,在两个实施例中实施10分钟的等待时间。随后立即记录反应器温度(ti-bd)并开始丁二烯(246.1g)进料操作。在2分钟内完成丁二烯装载。丁二烯聚合热升高了反应物的温度,并且当它达到55.8±0.3℃(tsicl4)时,将四氯化硅偶合剂射料(shot)进给至反应器中。四氯化硅(sicl4)的剂量示于表20中。记录直至偶合剂喷射(tsicl4)的丁二烯反应时间,从丁二烯进料操作开始计算。在sicl4喷射后6分钟,将羟基当量超过最初添加至反应器中的正丁基锂的醇溶液射料进给至反应器中。在这些实施例中,环己烷与总单体进料的比率为9.0。工艺参数(ti、tpst、tpst、tibd、tsicl4、tsicl4)列于表21中。来自该实施例的产物的sbs共聚物表征示于表22和表23中。使用现有技术的比较实施例c13和c14。进行与实施例15和16相同的聚合程序,但极性改性剂体系缺少dthfp。活性引发剂含量、极性改性剂与引发剂的摩尔比以及氯化硅剂量示于表20中。工艺参数(ti、tpst、tpst、tibd和tsicl4)列于表21中。来自该实施例的产物的sbs共聚物表征示于表22和表23中。表20,根据本发明的实施例15和16以及使用现有技术的比较实施例c13和c14中的引发剂、极性改性剂体系和偶合剂装料。表21.根据本发明的实施例15和16以及使用现有技术的比较实施例c13至c14中的聚合工艺参数。表22.根据本发明的实施例15和16以及使用现有技术的比较实施例c13和c14中产生的sbs共聚物的nmr表征。表23.根据本发明的实施例15和16以及使用现有技术的比较实施例c13至c14中产生的sbs共聚物的gpc表征。实施例mpuc,kg/mol偶合的sbs共聚物,wt%15119.779.916111.985.3c13127.144.7c14118.151.5mpuc=未偶合部分的峰值分子量。图5.根据本发明的实施例15和使用现有技术的比较实施例c13中产生的共聚物的分子量分布。当相对于使用如比较实施例c13和c14中基于极性改性剂体系bdmaee/smt的现有技术时的此类参数,对比用实施例15和16中使用的本发明极性改性剂dthfp/bdmaee/smt获得的偶合效率和分子量分布时,可证明,本发明极性改性剂体系比最接近的现有极性改性剂体系向远远更高量的聚合物阴离子直至丁二烯消耗结束提供活性。针对如果当发生sicl4装料时,每个聚合物阴离子都保持活性,则sicl4装料的理论最大偶合效率为95%至96%来计划sicl4装料。在比较实施例c13和c14中,约一半的聚合阴离子不能参与偶合反应,确认约一半的聚合阴离子在sicl4装料之前已终止。值得注意的是,这些实施例的环己烷、苯乙烯和丁二烯装料与实施例3至11以及比较实施例c7和c8的环己烷、第一苯乙烯和丁二烯装料一致。因此,实施例15和16中证明的直至丁二烯消耗结束的聚合物阴离子的活性适用于实施例3至11,所述实施例3至11共享优选的极性改性剂体系dthfp/bdmaee/smt,并获得单峰窄分子量分布的未偶合sbs产物。另一方面,比较实施例c13和c14中证明的聚合物阴离子的相同程度的过早终止适用于比较实施例c7和c8,它们全部使用最接近的现有技术体系bdmaee/smt,并显示出宽的双峰分子量分布。实施例17(假设).使用本发明的优选极性改性剂体系合成的实施例2的线性sbs嵌段共聚物,进一步氢化以获得线性高乙烯基sebs。通过以醇/li活性物=0.5的摩尔比添加2-甲基-2,4-戊二醇,使来自实施例2的展现87.6wt%总乙烯基含量且仍然溶解于环己烷(溶剂与聚合物的比率为8.0,w/w)中的线性sbs失活。此后,将聚合物溶液加热至90℃并添加每100g聚合物0.25mmol氢化催化剂(cp2ti(phoch3)2或cp2ti(ch2pph2),如us5985995a中所述),之后添加氢气直至达到8kg/cm2的压力。在45分钟内完成氢摄取。1hnmr分析显示其获得具有99.6%氢化的sebs。因此,由此获得衍生自具有87.6wt%乙烯基含量的sbs的新型线性sebs。实施例18(假设).使用本发明的优选极性改性剂体系合成的实施例10的线性sbs嵌段共聚物,进一步氢化以获得线性高乙烯基sebs。通过以醇/li活性物=0.5的摩尔比添加2-甲基-2,4-戊二醇,使来自实施例10的展现90.1wt%总乙烯基含量且仍然溶解于环己烷(溶剂与聚合物的比率为7.5,w/w)中的线性sbs失活。此后,将聚合物溶液加热至90℃并添加每100g聚合物0.25mmol氢化催化剂(cp2ti(phoch3)2或cp2ti(ch2pph2),如us5985995a中所述),之后添加氢气直至达到8kg/cm2的压力。在45分钟内完成氢摄取。1hnmr分析揭示其获得具有99.6%氢化的sebs。因此,由此获得衍生自具有90.1wt%乙烯基含量的sbs的新型线性sebs。实施例19(假设).使用本发明的优选极性改性剂体系合成的实施例15的星型sbs嵌段共聚物,进一步氢化以获得星型高乙烯基sebs。通过以醇/li活性物=0.5的摩尔比添加2-甲基-2,4-戊二醇,使来自实施例15的展现89.4wt%总乙烯基含量且仍然溶解于环己烷(溶剂与聚合物的比率为8.0,w/w)中的星型sbs嵌段共聚物失活。此后,将聚合物溶液加热至90℃并添加每100g聚合物0.25mmol氢化催化剂(cp2ti(phoch3)2或cp2ti(ch2pph2),如us5985995a中所述),之后添加氢气直至达到8kg/cm2的压力。在45分钟内完成氢摄取。如通过1hnmr分析所揭示的,获得具有99.6%氢化的星型sebs。因此,由此获得衍生自具有89.4wt%乙烯基含量的星型sbs的新型星型sebs。衍生自高乙烯基含量的sbs的来自实施例17至19的sebs是必定与已知sebs相比与聚丙烯更相容的新型材料。实施例20(假设).使用本发明的优选极性改性剂体系合成的实施例10的线性sbs嵌段共聚物,进一步进行末端官能化并进一步进行氢化以获得线性高乙烯基末端官能化的sebs-f。通过添加定量的氧化丙烯或n-亚苄基甲胺(官能化剂/li活性物的摩尔比=1.0),用oh或仲胺官能团将来自实施例8的展现90.1wt%的总乙烯基含量且仍然溶解于环己烷(溶剂与聚合物的比率为8.0,w/w)中的线性sbs末端官能化。此后,将聚合物溶液加热至90℃并添加每100g聚合物0.25mmol氢化催化剂(cp2ti(phoch3)2或cp2ti(ch2pph2),如us5985995a中所述),之后添加氢气直至达到8kg/cm2的压力。在45分钟内完成氢摄取。如通过1hnmr分析所揭示的,获得具有99.6%氢化的末端官能化sebs-f。因此,由此获得衍生自具有90.1wt%乙烯基含量的线性sbs的新型线性sebs-oh或sebs-nhr。来自实施例20的末端官能化sebs是展现oh或胺极性基团与沿着中心eb嵌段富集高丁烯的组合的新材料。这些新型材料可用作反应性聚合物,以实现可用于生产新型嵌段共聚物的链延伸反应,并可被更有效地用于工程塑料冲击改性和聚合物共混物增容。本发明的实施方案实施方案1.通过有机锂引发的阴离子聚合制备乙烯基芳族单体和共轭二烯单体的嵌段共聚物的极性改性剂体系,其包含:(a)具有式(i)结构的化合物:其中r1至r14独立地为-h或-cnh2n+1基团,其中n=1至6,并且其中双四氢糠基丙烷(dthfp)是具有式(i)结构的优选化合物。(b)具有式(v)结构的化合物:其中m=1至2,n=1至2;并且其中r1至r4独立地为-cxh2x+1基团,其中x=1至6,并且其中双[2-(n,n-二甲基氨基)乙基]醚是具有式(v)结构的优选化合物;和(c)醇钠化合物,优选薄荷醇钠,其中双四氢糠基丙烷与所述有机锂引发剂的摩尔比在约0.36:1至约4.2:1、优选约1.5:1至约3.5:1且更优选约2.5:1至约3.0的范围内;其中双[2-(n,n-二甲基氨基)乙基]醚与所述有机锂引发剂的摩尔比在约0.1:1至约1.5:1、或约0.2:1至约1:1、优选约0.3:1至约1:1、更优选约0.4:1至约0.9:1的范围内;其中醇钠化合物与所述有机锂引发剂的摩尔比在约0.01:1至约0.3:1、或约0.02:1至约0.2:1、或优选约0.03:1至约0.15:1、或更优选约0.04:1至约0.10:1的范围内。实施方案2.通过有机锂引发的阴离子聚合制备乙烯基芳族单体和共轭二烯单体的嵌段共聚物的极性改性剂体系,其包含:(a)具有式(ii)结构的化合物:其中r1是-cnh2n+1基团,其中n=1至6;并且其中r2至r7独立地为-h或-cxh2x+1基团,其中x=1至6。(b)具有式(v)结构的化合物:其中m=1至2,n=1至2;并且其中r1至r4独立地为-cxh2x+1基团,其中x=1至6,并且其中双[2-(n,n-二甲基氨基)乙基]醚是具有式(v)结构的优选化合物;和(c)醇钠化合物,优选薄荷醇钠,其中乙基四氢糠基醚与所述有机锂引发剂的摩尔比在约0.3:1至约4:1、优选约1:1至约3:1且更优选约1.5:1至约2.5:1的范围内,其中双[2-(n,n-二甲基氨基)乙基]醚与所述有机锂引发剂的摩尔比在约0.1:1至约1.5:1、或约0.2:1至约1:1、优选约0.3:1至约1:1、更优选约0.4:1至约0.9:1的范围内;其中醇钠化合物与所述有机锂引发剂的摩尔比在约0.01:1至约0.3:1、或约0.02:1至约0.2:1、或优选约0.03:1至约0.15:1、或更优选约0.04:1至约0.10:1的范围内。实施方案3.通过有机锂引发的阴离子聚合制备乙烯基芳族单体和共轭二烯单体的嵌段共聚物的极性改性剂体系,其包含:(a)具有式(iii)结构的化合物:其中r1至r4独立地为-ch3或-ch2ch3基团;并且其中r5是-h或-cnh2n+1基团,其中n=1至2,并且其中n,n,n’,n’-四甲基乙二胺是具有式(iii)结构的优选化合物。(b)具有式(v)结构的化合物:其中m=1至2,n=1至2;并且其中r1至r4独立地为-cxh2x+1基团,其中x=1至6,并且其中双[2-(n,n-二甲基氨基)乙基]醚是具有式(v)结构的优选化合物;和(c)醇钠化合物,优选薄荷醇钠,其中n,n,n’,n’-四甲基乙二胺与所述有机锂引发剂的摩尔比在约1:1至约4:1、优选约1.5:1至约3:1且更优选约2:1至约3:1的范围内,其中双[2-(n,n-二甲基氨基)乙基]醚与所述有机锂引发剂的摩尔比在约0.1:1至约1.5:1、或约0.2:1至约1:1、优选约0.3:1至约1:1、更优选约0.4:1至约0.9:1的范围内;其中醇钠化合物与所述有机锂引发剂的摩尔比在约0.01:1至约0.3:1、或约0.02:1至约0.2:1、或优选约0.03:1至约0.15:1、或更优选约0.04:1至约0.10:1的范围内。实施方案4.通过有机锂引发的阴离子聚合制备乙烯基芳族单体和共轭二烯单体的嵌段共聚物的极性改性剂体系,其包含:(a)具有式(iv)结构的化合物:其中r1至r6独立地为-h或-cnh2n+1基团,其中n=1至6;其中r7至r8独立地为-cxh2x+1基团,其中x=1至2,并且其中四氢糠基-n,n-二甲胺是具有式(iv)结构的优选化合物。(b)具有式(v)结构的化合物:其中m=1至2,n=1至2;并且其中r1至r4独立地为-cxh2x+1基团,其中x=1至6,并且其中双[2-(n,n-二甲基氨基)乙基]醚是具有式(v)结构的优选化合物;和(c)醇钠化合物,优选薄荷醇钠,其中四氢糠基-n,n-二甲胺与所述有机锂引发剂的摩尔比在约1:1至约4:1、优选约1.5:1至约3:1且更优选约2:1至约3:1的范围内,其中双[2-(n,n-二甲基氨基)乙基]醚与所述有机锂引发剂的摩尔比在约0.1:1至约1.5:1、或约0.2:1至约1:1、优选约0.3:1至约1:1、更优选约0.4:1至约0.9:1的范围内;其中醇钠化合物与所述有机锂引发剂的摩尔比在约0.01:1至约0.3:1、或约0.02:1至约0.2:1、或优选约0.03:1至约0.15:1、或更优选约0.04:1至约0.10:1的范围内。上文已描述了本发明,技术、程序、材料和设备的各种修改对于本领域技术人员来说将是显而易见的。意在将本发明的范围和精神内的所有此类变化都包括在所附权利要求的范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1