用于在染色体上原位杂交的DNA探针的制作方法
本发明涉及产生用于诊断染色体畸变的染色体原位杂交用探针的方法,以及由此产生的dna探针和探针混合物。
背景技术:
通常可在肿瘤细胞中观察到染色体畸变。可以通过g显带或者通过用于特定基因组基因座的探针进行原位杂交(ish)来确定染色体畸变。在多色荧光原位杂交中,以不同的颜色对基因组的几个区域进行标记;这些区域也可以位于距离疾病的基因座和断裂点一定距离处,因此即使是复杂的染色体畸变也可以很容易地被诊断出来。一些畸变仅发生在特定的肿瘤中(例如费城染色体伴有某些白血病);其他染色体畸变示出治疗类型,特别是对乳房肿瘤的治疗。本文中,临床上相关的是编码细胞表面受体的致癌基因erbb2和染色体17的着丝粒区域的cen17区域。ish提供了评价肿瘤侵入性和对于患者而言更有针对性的治疗的手段。还可以通过遗传变异来区分非霍奇金淋巴瘤的亚组。
产生ish探针的常规方法使用bac克隆(细菌人工染色体)、yac克隆、粘粒和fos-质粒(fosmid)。它们包含基因组dna的大区域(高达50万个碱基),因而除了所寻找的序列之外还包含重复序列、假基因和旁系同源序列。这些物质在显色或荧光ish中造成非特异性杂交和背景,这使得评价更为困难并且有时变得不可能。特别是在cish(显色原位杂交)中,通常难以避免高背景。cish探针通常比fish(荧光原位杂交)探针小。
为了避免由重复序列造成的背景,在bac探针的情况下经常添加大过量的所谓封闭dna(例如cot-1dna、鲑鱼精子dna、trna等等)。然而,仍然不能完全避免来自重复序列的信号。因此,通常还尝试“释放”重复序列的bac克隆;参见swennenhuisjf等人,constructionofrepeat-freefluorescenceinsituhybridizationprobes,2011,nucleicacidsresearch,2012,第40卷,no.3,e20,doi:10.1093/nar/gkr1123;wo2007/053245和图2中的现有技术。bac克隆是随机片段化的,并且片段配置有接头并在pcr中扩增。添加过量的cot-1dna后,使dna溶解并在65℃下用双链特定核酸酶(dsn-双链特定核酸酶)进行消化。将cot-1dna存在下的扩增和消化步骤重复数次,直至得到“无”重复人序列的探针或探针混合物。也可以通过其他方法实现重复序列的去除;参见craidjm等人,removalofrepetitivesequencesfromfishprobesusingpcr-assistedaffinitychromatography,humangenetics(springer,berlin,de)第100卷,第472-476页;us2004029298、wo2004/083386;wo01/06014。但是这些方法不仅要付出相当大的努力,而且遗憾的是,这些方法也并不完整或有把握。也不可能简单地将探针dna固定在质粒中。
也可以在pcr(聚合酶链式反应)中生产ish探针。pcr发生在基因组上,特别是在无重复序列的区域上,并且通常仅编码基因或结构域的一部分。在人类基因组各处和所有染色体上都可以找到重复序列和alu序列。us8,407,013b2公开了计算机辅助序列分析和通过pcr从头开始生成基因组探针;参见roganpk等人sequence-baseddesignofsingle-copygenomicdnaprobesforfluorescenceinsituhybridization,genomeres.(2001)11(6):1086-1094。关于现有技术,还可以参见us6150160-a、us6828097b1、us7014997b2、us20003002204a1、ep1127163、us200030108943a1、de69032920、us200030194718a1、us200040161773a1、wo2004/04097050、wo2004/083386、ep1285093、wo2014/036525-a1、wo2000/188089、ep2069537、ep1127163、ep1669902以及根据de102010029855a1的方法。然而,无重复序列的探针的产生仍然存在问题,因为这些类型的探针的覆盖范围通常不大,可是临床相关的ish探针需要几百万个碱基。
此外,探针还必须用放射性基团、显色基团或荧光基团进行标记。可以通过切口平移反应、通过随机引物法或通过直接使用经标记的核苷酸进行pcr标记的方式酶促地进行标记和/或通过化学偶联来进行标记。然而,扩增反应中的标记是复杂的,因为必须为每个标记、每种荧光染料和每个显色基团重新建立聚合酶链式反应。扩增反应中的标记还取决于序列长度、修饰的化学结构、标记和核苷酸之间的接头长度,以及聚合酶和起始材料的条件。因此,现有技术存在问题。
技术实现要素:
通过根据权利要求1的方法和根据权利要求13和14的探针解决了该问题。该方法和探针的有利实施方案可以参见从属权利要求,并在实施例中对其进行描述。
该方法包括产生直接或间接标记的核酸,该方法包括在较大的基因组区域中分析具有特定序列的区段的序列以及对于特定基因座选择特定核酸序列;在选定的特定核酸序列上设计并合成用于聚合酶链式反应的有义和反义引物对,其中合成的引物各自包含与非特定核酸序列的链或互补链互补的序列以及相同的非互补接头序列,该相同的非互补接头序列在严格条件下不与基因组杂交,并且可以兼性地含有用于限制性核酸内切酶的切割序列;具有若干有义和反义引物对的若干第一聚合酶链式反应,并且在对反应产物进行合并后,得到包含已知(非重复)序列的合成的pcr片段的第一混合物(池a);利用引物对合成的pcr片段的混合物(池a)进行多重聚合酶链式反应,所述引物在非互补的接头序列上杂交,并得到具有扩增的序列可控的pcr片段的混合物(池b),该pcr片段无重复部分,然后,上述具有扩增的序列可控的pcr片段(单独地或在混合物中)适合用于染色体的显色原位杂交或荧光原位杂交(cish或fish)。
在一个实施方案中,在第一聚合酶链式反应之后,对混合物中存在的合成的核酸片段进行大小选择性分析,然后进行纯化。此外,在最后的扩增步骤中添加经修饰的或经标记的核苷酸(pcr标记)是有利的。如果在最后的扩增步骤中添加已经被修饰的核苷酸,则所述核苷酸可以为能够与显色基团或荧光基团进行化学偶联的类型,优选为氨基烯丙基ntp。
在另一个实施方案中,将由第一聚合酶链式反应所产生的核酸片段克隆到质粒中。专业技术人员将认识到这可以在接头序列的限制下完成。在质粒的扩增之后,可以通过接头以任意数量产生片段。
探针片段还可以进行这样的反应,该反应将报告基团插入或附着到杂交探针中/上。插入的标记可以是放射性标记、显色标记或荧光标记。显色基团包括半抗原,如生物素、抗生物素蛋白、地高辛,因为可以以已知的方式利用经标记的抗体使这些半抗原在免疫反应中可见。其他显色基团为能够催化显色反应的酶,如过氧化物酶或乳糖酶。还可以使用具有反应性基团的经修饰的核苷酸(如烯丙胺),其可以与适当的染料基团进行反应。
本公开的方法的优点在于,可以对步骤(a)中选定的非重复核酸序列进行选择,使其在第一多重聚合酶链式反应中以基本相同的频率扩增。优选在步骤(a)中选择序列区段,使得非重复pcr片段具有100至5,000个碱基对,优选具有100至1,000个碱基对。特别优选的是具有400至600个碱基对的片段。此外,如果在进行分析时,在分析步骤中选定的非重复核酸序列在基因组上彼此相邻,从而由原位杂交产生更高的信号强度则是有利的。
通常需要几种具有不同标记的探针来检测染色体畸变。因此,本公开还包括生产大量具有不同标记的探针。如果在第一步骤中选定的特定核苷酸序列与染色体中的断裂点区域相邻则是有利的。对于诊断目的而言特别有利和实用的是,当在分析步骤中选定的特定序列位于断裂点区域的侧翼并且具有不同的标记时,由于染色体畸变从而可以直接地诊断。还可以为相邻序列选择不同的探针标记,使得颜色着色剂最初产生复合颜色,并且在畸变的情况下可以观察到颜色变化或两种不同的颜色信号。也可以进行相反的处理,即,如果存在畸变,则两种不同的颜色信号可以产生复合信号或融合信号。在其他情况下,根据需要,还可以选择在畸变的情况下被扩增的来自单个区域的序列或者在该区域侧翼的序列作为平衡易位、不平衡易位和相互易位的一部分。
探针的标记优选选自以下物质的组:显色分子、聚甲炔染料、噻唑和噁唑染料、hoechst33342(2'-(4-乙氧基苯基)-5-(4-甲基-1-哌嗪基)-2,5'-双-1h-苯并咪唑三盐酸盐)、4',6-二脒-2-苯基吲哚、alexa405、alexa488、alexa594、alexa633;德克萨斯红、罗丹明;磺化和非磺化花青染料、cy2、cy3、cy5、cy7;荧光分子、荧光素、5,6-碳氟荧光素、fitc(异硫氰酸荧光素)、gfp(绿色荧光蛋白);化学发光分子、吖啶;
一个实施方案涉及提供用于原位杂交以检测染色体畸变的经标记的探针,该探针包含多个pcr片段,所述pcr片段的序列不包含任何重复序列、假基因或旁系同源基因,并且在人类基因组上彼此相邻。另一个实施方案涉及用于特定染色体畸变的探针混合物或检测试剂盒,其包含位于特定断裂点区域侧翼的几种经不同标记的探针。
在例子中并参照所附图示描述了本发明的其他优点、实施方案和优点。
附图说明
以下图示出:
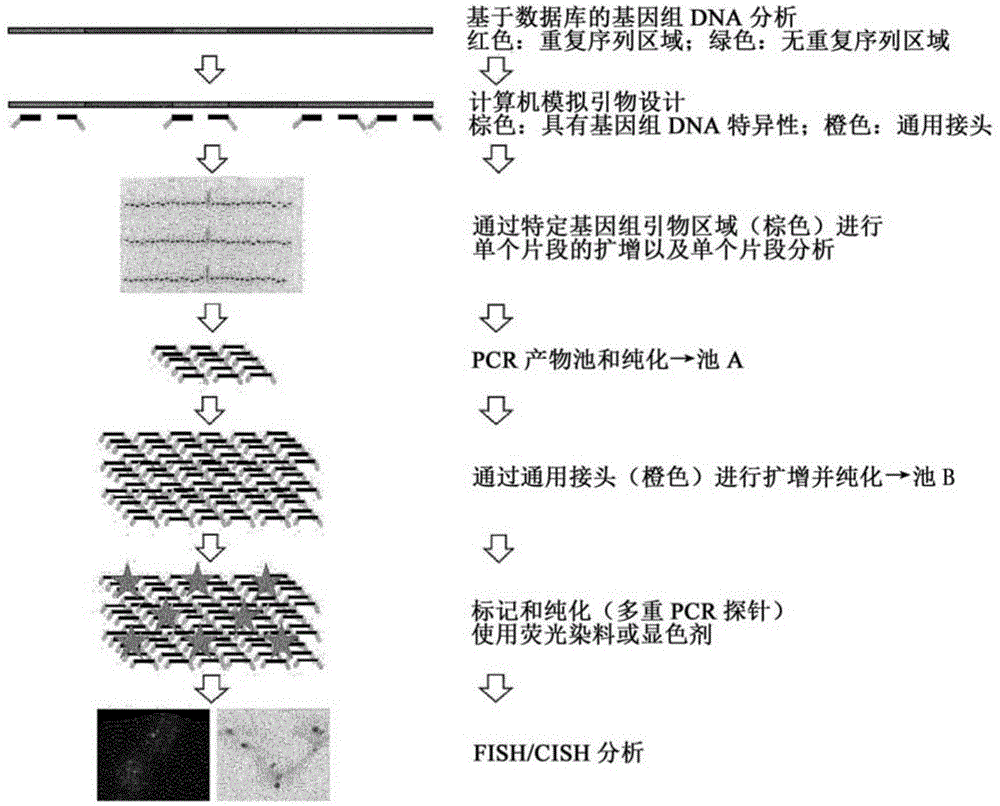
图1:产生用于染色体原位杂交(ish)的序列可控的pcr探针的步骤图;
图2:根据现有技术得到重复减少的ish探针的步骤的示意图;
图3:利用dapi(4',6-二氨基苯酚吲哚)对细胞核进行可控染色和各种原位杂交之后的样品的荧光显微镜图像,其中在pcr反应中对序列可控的ish探针进行标记(“一步法”):左列:利用dapi对细胞核进行可控染色;右列:利用探针对her2、mdm2、met和fgfr1基因或着丝粒进行原位杂交;
图4:利用dapi(4',6-二氨基苯酚吲哚)对细胞核进行可控染色和各种原位杂交之后的样品的荧光显微镜图像,其中通过切口平移的方式对序列可控的ish探针进行标记:左列:利用dapi对细胞核进行可控染色;右列:利用探针对her2、mdm2、met和fgfr1基因或着丝粒进行原位杂交。
具体实施方式
可以由基因组dna产生序列可控的pcr探针。人类基因组的全序列是已知的并且可以从数据库获得。其为fish/cish探针设计的起点。如果载体及其序列可用,也可以从bac克隆或其他dna载体(质粒、粘粒、fos-质粒、yac)的序列开始。也可以使用其他载体进行下面描述的基因组dna的所有内容。
参见图1,在第一步骤(a)中,使用数据库中的序列通过计算机分析研究该区域的基因组序列和遗传组成。在这种情况下,在基因组序列中寻找并鉴定具有简单重复序列、alu序列和复杂重复序列以及所有假基因和旁系同源区段的所有区域。这些序列不适合用于ish探针。ncbi(国家生物技术信息中心,马里兰州贝塞斯达)或ensembl(维护方为欧洲生物信息学研究所(ebi)和欧洲分子生物学实验室(embl),德国海德堡)提供了公共基因组数据库,或者可以通过ucsc基因组浏览器(美国加州大学,圣克鲁兹分校,美国)获取公共基因组数据库。可以通过常规分析程序进一步缩小并鉴定任何非特定序列区域。
杂交探针中包含仅特异于基因座并且仅出现一次的序列。对在染色体杂交期间在其他基因座处杂交从而非特异性地对背景作出贡献的序列进行排除(步骤a)。然后,对于以这种方式控制的在染色体中仅出现一次的基因组序列,创建有义和反义引物文库(b)。利用特定引物序列和通用接头合成各引物。基于已知的基因组序列来设计和选择有义和反义引物,使得pcr和随后的扩增产生长度基本上相同的dna片段。通常,为每个基因组区域设计并合成200个引物,使得在pcr之后,基因组上的基因座被100个dna片段逐段覆盖。在第一扩增中,借助于引物在单独的pcr反应中产生特定片段;检查所得的pcr片段的纯度和大小,并在步骤(d)中进行组合或合并(池a)。在对池(池a)进行纯化之后,将10ng合并的pcr-dna用作第二扩增的模板(步骤e),其中接头在该扩增中用作有义和反义引物。由于池a的所有片段具有相同的接头和设计的相似大小,因而来自池a的所有pcr片段在该反应(多重pcr)中被扩增至相同长度。结果得到pcr片段的混合池(池b)。
如果在该最后的反应中添加经标记的核苷酸(一步法pcr标记),则结果得到具有经标记的pcr片段的池b。纯化后,其可用于原位杂交。或者,也可以在第二扩增之后通过切口平移对含有混合pcr片段的池b进行标记。
为了在fish/cish中稳定地检测基因座,通常需要研究10万至100万个碱基的基因组区域用于探针设计。这意味着会产生1至10个多重pcr探针,其组合构成实际探针。由此产生的探针从其产生开始时就不含重复元素、假基因或旁系同源物。可以产生任意数量的探针。由于单个pcr片段的大小基本上相似,或者pcr片段的长度是特定的,因此将经标记的核苷酸直接插入不再是非常苛刻的过程。在通常的多重pcr或接头pcr中,可以使用以下物质:(a)当通过常规酶促方法(如切口平移)进行后续标记时,使用非化学修饰或未修饰的dntp,(b)使用荧光染料(例如atto488或cy3)或半抗原(例如地高辛、二硝基苯酚或生物素)标记的dntp,其中可以直接在接头pcr中进行标记。关于用荧光染料或半抗原标记的dntp的添加,有必要确定其添加比例。根据标记和dntp(dutp:荧光标记的dntp),该比例将为1:1至1:20;或(c)化学反应性dntp,如氨基烯丙基dntp。也可以在合成之后通过与胺反应性nhs酯活化的(nhs=n-羟基琥珀酰亚胺)染料或半抗原衍生物的化学偶联进行标记。根据探针类型(基因探针或着丝粒探针),以5:1至1:5的比例添加反应性氨基烯丙基核苷酸或氨基烯丙基dntp。然后通过与胺活性染料或荧光染料的反应在合成后对dna探针进行标记。如果不直接使用荧光染料或半抗原标记dna探针,则必须在接头扩增后进行标记。
实施例
实施例1her2基因的基因座探针的产生
1.her2基因座探针的计算机模拟(insilico)设计
在染色体17上,选择具有her2基因和大的5'和3'侧翼的基因组区域用于her2(erbb2)基因探针的设计。her2基因探针覆盖染色体17上从39,395,605至39,799,506的区段。在ensembl(www.ensembl.org)和ncbi(https://www.ncbi.nlm.nih.gov/)数据库中对基因组序列进行分析,并且为探针鉴定四个合适的区段。第一基因组区段为染色体17的39,395,605至39,569,361。以下说明仅涉及该区段;对her2基因座处的三个其他基因组区段进行类似处理。
利用常规分析程序鉴定基因组区段39,395,605至39,569,361中的所有重复元素、alu序列、假基因和旁系同源序列区段。排除重复元素、假基因和旁系同源序列。将对第一基因组区段具有特异性的序列分成具有250至800个碱基对的区段。不考虑具有少于250个碱基对的部分序列。在该分析之后,四个选定的基因组区段中的每一个中的靶基因组序列仅含有起始序列的40%至60%,从而使大约一半的基因组起始序列作为非特异性的而被排除在外。
2.计算机模拟引物设计
针对具有250至800个碱基对的基因组片段,设计并合成用于特定序列区段的有义和反义引物。一方面,引物与待扩增的基因组序列互补;另一方面,引物还包含具有用于限制性核酸内切酶的切割位点的通用接头序列。设计有义和反义引物,使得在基因组的pcr中的得到的产物具有相似的长度。理想的片段长度为大约500个碱基对。只有在引物对的特异性或功能性需要时才会出现偏差。为四个基因组区段中的每一个设计并合成了大约200个引物。说明书所附的表1包含由此确定的染色体17上的基因组区段39,395,605至39,569.361的有义和反义引物的代表性列表。
3.单个片段的扩增
对于每个有义和反义引物对,在50μl溶液中扩增基因组片段。使用高通量法(96孔)进行单个pcr反应,每种情况下,退火温度(引物退火)为55℃、15秒链延伸,进行超过35个循环。在琼脂糖凝胶上检查所得的pcr片段的纯度和大小。仅使用具有预期大小的特定条带的pcr产物,以便有效控制和得知其序列。合适的pcr产物的收率超过90%。
4.利用限制性酶切割单个片段并进行克隆
通常将pcr产物直接克隆到载体质粒中而不进行切割,从而得到固定。一些pcr产物也会在切割后被克隆到质粒中。分开进行的测序检查可以以简单的并且以已知的方式进行。
5.单个片段的合并和处理
对大约100个pcr产物进行合并及处理,以除去引物、蛋白质、游离核苷酸和盐。然后通过琼脂糖凝胶电泳对混合物(池a)进行光度测量和光学分析。
6.通过通用接头(多重pcr)进行池的扩增并进行标记
为了得到高产率,直接使用单个片段的混合物(池a)作为模板,使用高效taq聚合酶进行通过通用接头的第二扩增(多重pcr)。将用荧光染料(例如atto488或cy3)标记的dntp用于该多重pcr/接头pcr。以测试比例添加荧光标记的dntp,所述测试比例取决于经标记的dntp,并且根据探针的类型(基因探针或着丝粒探针)而不同。不同探针之间的比例在5:1和1:5之间,并且取决于标记。
7.经标记的dna探针的纯化
标记和扩增反应之后,通过沉淀和层析对经标记的多重pcr探针进行最终纯化,其中除去游离dntp、经标记的核苷酸、有义和反义引物以及酶。然后进行光度测量并测定fish探针中荧光的插入率并通过琼脂糖凝胶电泳对探针进行最终检查。
8.dna探针的额外片段化
作为进一步的选择,如果dna探针由于其长度而在ish中产生过多的背景,则对其进行后片段化。通常,对于ish而言200至300个碱基对的长度是有利的。后片段化可通过物理方法完成,但也可通过酶促方法或化学方法完成。通过琼脂糖凝胶电泳分析片段大小分布。
9.dna探针的完成
her2基因座探针包含四个区段。上述步骤1至8涉及第一区段。最终的dna探针由四个独立的制剂(4个多重pcr探针)组成。或者,虽然具有较少的控制,但可以在通用接头扩增之后对这四个单独的制剂进行组合,并一同进行标记和纯化。根据基因座、探针类型(扩增探针、断裂探针、融合探针)和用户说明,dna探针混合物将由1至10个制剂(多重pcr探针)组成。
实施例2染色体与经标记的pcr探针的原位杂交
版本1:例如根据dgho(德国血液学和内科肿瘤学会)的实验室工作组的建议,可以使用标准方法在组织或细胞上进行与dna探针的ish。在这种情况下,原位杂交发生在细胞培养物或组织的间期细胞上。在每种情况下,靶dna为间期细胞核的核dna,将其固定在样品载玻片上。如实施例1中所述产生和标记本文的探针。通常使用荧光染料dapi(4',6-二脒基-2-苯基吲哚)对细胞核进行复染。原则上,可以在以下材料上进行fish:外周血(pb)、骨髓(km)、石蜡切片、肿瘤组织、细胞离心涂片制剂、羊水、用甲醇冰醋酸固定的细胞/中期等。诸如血液和骨髓之类的其他患者样品在ficoll分离后,用甲醇/乙酸(比例3:1)进行固定,并在-20℃冷冻直至杂交。羊水或石蜡切片需要特殊的预处理。
版本2:在实施例中(参见图3和图4),根据标准方法在超薄切片机上对包埋在石蜡中的细胞系(细胞培养物)的细胞进行切割,并将其捞至清洁的样品载玻片上。随后根据标准方法对包埋在石蜡中的细胞进行处理。通过一系列二甲苯和醇系列的分级洗涤从而确保脱蜡。在95℃下,在柠檬酸盐预处理缓冲液中进行仅几分钟的预处理。随后用胃蛋白酶溶液对细胞进行部分消化,从而尽可能多地从细胞质中释放细胞核并使其可用。在连续的酒精洗涤中脱水后,将探针-杂交混合物移液到细胞上并用盖玻片和密封剂进行密封。在75℃下将细胞核和杂交样品变性10分钟,缓慢冷却至37℃并在37℃的潮湿腔室中杂交过夜(16小时)。除去盖玻片中之后,在37℃下用冲洗液进行两次冲洗。利用合适的滤光片在荧光显微镜上评价和记录杂交信号。图3和图4描绘了以不同方式标记的探针(通过切口平移或一步法pcr标记)的结果。在两种情况下,杂交信号都清晰可见,并且背景杂交可忽略不计。
表1
染色体17上的基因组区段39,395,605至39,569,361的有义和反义有义和反义引物
- 还没有人留言评论。精彩留言会获得点赞!