通过杂交找到低丰度序列(FLASH)的方法与流程
本申请主张2016年8月16日提交的美国临时申请序列号62/375,789的权益,该申请通过引用并入文中。
背景技术:
目前用于富集复杂核酸文库中的低丰度序列的方法通常涉及多重pcr或与标记的寡核苷酸的杂交。这两种方法效率低,难以实施,优化成本高,并且在给定样品中可以富集的序列数量有限。
一直需要用于富集核酸样品中低丰度序列的新颖方法。
技术实现要素:
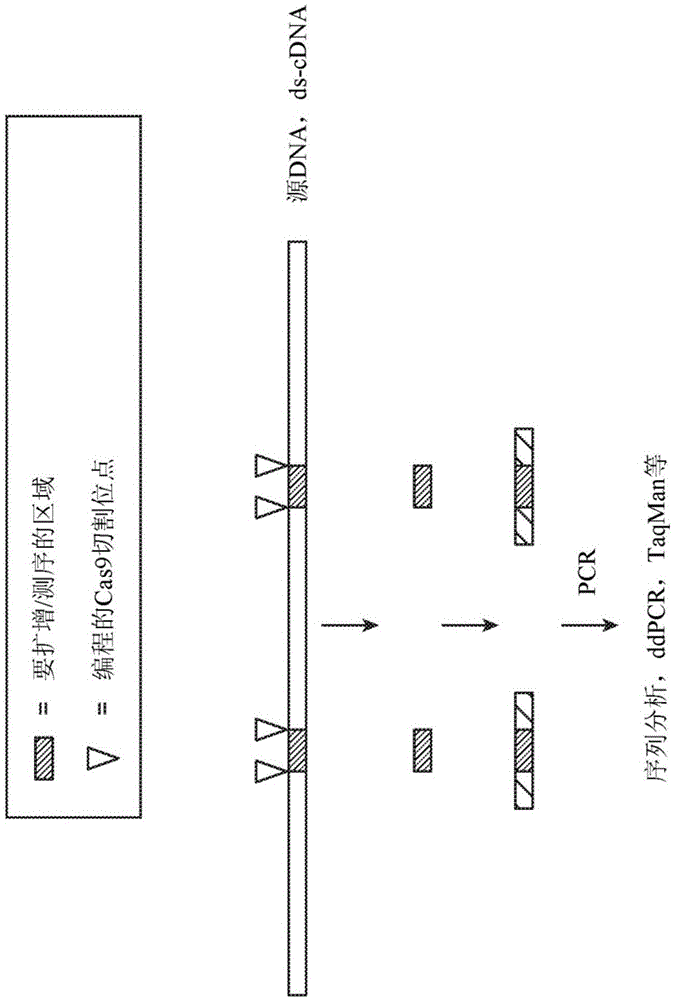
本文描述的是称为通过杂交找到低丰度序列或“flash”的方法,一种在测序或其它分子计数应用前使用序列特异性核酸酶(例如crispr/cas9)来切割dna文库或其它样品中的特定目标位点的技术。在一些实施中,然后将新暴露的dna末端自由连接到允许他们被扩增的特定衔接序列。在这些实施例中,仅使用对衔接子特异的一对引物的单个pcr步骤因此可以以完全可编程的方式扩增数百、数千或可能数百万种不同的序列。在一些情况下,为了减少非靶向分子的测序,可以封端dna来源中dna分子的末端,例如用磷酸酶处理或在核酸酶消化之前使用另一种方法来封端任何已经可及的dna末端。
在一些实施例中,所述方法可以包括:(a)用多种重编程的核酸指导的内切核酸酶消化末端封端的(例如,磷酸酶处理的)混合核酸样品,所述内切核酸酶靶向目标序列以产生消化的样品,其中消化的样品中的至少一些片段包含:(i)目标序列和(ii)至少一个通过内切核酸酶切割产生的可连接末端;(b)富集含有目标序列的片段;以及(c)分析富集的片段。
还提供了用于执行所述方法的试剂盒。
附图说明
当结合附图阅读时,将最好地理解以下详细描述的某些方面。需要强调的是,根据惯例,附图的各种特征未按比例绘制。相反,为了清楚起见,各种特征的尺寸被任意扩大或缩小。附图中包括以下图:
图1显示了flash方法的一些原理。
图2显示了不同的flash池(由空心和实心三角形表示)如何用于将具有重叠片段的所需dna片段(实心条)片段化。可以使用不同的flash池(由空心和实心三角形表示)来将具有重叠片段(散列条)的所需dna片段(实心条)片段化。重叠区域可以靶向snp、易位或侧翼重复序列的区域。这些重叠片段可以在文库制备之前组合,然后用于组装测序读数。
图3图a显示来自i)甲氧西林敏感性金黄葡萄球菌(mssa)的培养分离物,ii)耐甲氧西林金黄色葡萄球菌(mrsa)和iii)直接来自mrsa肺炎患者的bal液的meca丰度。来自mrsa肺炎患者的bal液中可检测的meca随着flash而增加。面板b显示flash允许检测来自mrsa和铜绿假单胞菌肺炎患者的bal液中的氟喹诺酮抗性突变gyras84l。
图4描述了该方法的另一种形式,其中封端的衔接子与片段化dna连接,防止聚合酶扩增,除非靶向切口酶(例如具有单个活性位点突变d10a的cas9)产生允许置换衔接子的封端链的单链切割。结果是仅扩增含有两个切口酶位点的插入物。
定义
在更详细地描述示例性实施例之前,阐述以下定义以说明和定义说明书中使用的术语的含义和范围。
数字范围包括定义范围的数字。除非另有说明,否则核酸以5'至3'方向从左向右书写;氨基酸序列分别以氨基至羧基方向从左向右书写。
除非另外定义,否则本文使用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常所理解的相同的含义。singleton等人,dictionaryofmicrobiologyandmolecularbiology,第2版,johnwiley和sons,newyork(1994),以及hale&markham,theharpercollinsdictionaryofbiology,harperperennial,n.y.(1991)向技术人员提供本文使用的许多术语的一般含义。尽管如此,为了清楚和便于参考,下面定义了某些术语。
必须注意的是,如本文和所附权利要求中所使用的,单数形式“一(a)”,“一(an)”和“所述”包括复数指示物,除非上下文另有明确说明。例如,术语“引物”是指一种或多种引物,即单一引物和多种引物。还应注意,权利要求可撰写为排除任何任选元素。因此,此陈述旨在作为使用与权利要求元素的叙述相关的“单独”,“仅”等排他性术语或使用“否定”限制的先行基础。
如本文所用的术语“样品”涉及材料或材料混合物,通常但不一定是液体形式,其含有一种或多种目标分析物。本文使用的核酸样品可能是复合的,因为他们含有多种含有序列的不同分子。由来自哺乳动物(例如小鼠或人)的mrna制备的基因组dna和cdna是复合样品的类型。复合样品可具有超过104、105、106或超过107种不同的核酸分子。dna靶标可能来自任何来源,如基因组dna、cdna(来自rna)或人工dna构建体。文中可以使用任何含有核酸的样品,例如由组织培养细胞制成的基因组dna、组织样品、ffpe样品、临床、环境或其它类型的样品。
如本文所用的术语“核酸样品”表示含有核酸的样品。本文使用的核酸样品可以是复合的,因为他们含有多种含有序列的不同分子。来自哺乳动物(例如小鼠或人)的基因组dna、rna(和由其制备的cdna)是复合样品的类型。复合样品可具有超过104、105、106或超过107种不同的核酸分子。靶分子可能来自任何来源,例如基因组dna,或人工dna构建体。文中可以使用任何含有核酸的样品,例如由组织培养细胞制成的基因组dna或组织样品。
如本文所用,术语“混合物”是指散布而不以任何特定顺序的元素的组合。混合物是不均匀的,并且在空间上不能分离成其不同的组分。元素混合物的实例包括溶解在相同水溶液中的许多不同元素和在随机位置(即,没有特定顺序)附着于固体载体的许多不同元素。混合物不可寻址。为了举例说明,如本领域公知的,空间上分离的表面结合的多核苷酸阵列不是表面结合的多核苷酸的混合物,因为表面结合的多核苷酸的种类在空间上是不同的并且阵列是可寻址的。
术语“核苷酸”旨在包括那些不仅含有已知嘌呤和嘧啶碱基,还含有已经被修饰的其它杂环碱基的部分。这些修饰包括甲基化嘌呤或嘧啶、酰化嘌呤或嘧啶、烷基化核糖或其它杂环。此外,术语“核苷酸”包括那些含有半抗原或荧光标记的部分,并且不仅可以含有常规的核糖和脱氧核糖,还可以含有其它糖。修饰的核苷或核苷酸还包括对糖部分的修饰,例如,其中一个或多个羟基被卤素原子或脂族基团替换,被官能化为醚、胺等。
术语“核酸”和“多核苷酸”在本文中可互换使用以描述任何长度的聚合物,例如,大于约2个碱基、大于约10个碱基、大于约100个碱基、大于约500个碱基、大于约1000个碱基、至多约10,000个或更多个由核苷酸(例如脱氧核糖核苷酸或核糖核苷酸)组成的碱基,并且可以通过酶促或合成产生(例如,如美国专利号5,948,902和其中引用的参考文献中所述的肽核酸或pna),其可以以与两种天然存在的核酸类似的序列特异性方式与天然存在的核酸杂交,例如,可以参与watson-crick碱基配对相互作用。天然存在的核苷酸包括鸟嘌呤、胞嘧啶、腺嘌呤、胸腺嘧啶、尿嘧啶(分别为g、c、a、t和u)。dna和rna分别具有脱氧核糖和核糖糖骨架,而pna的骨架由重复的通过肽键连接的n-(2-氨基乙基)-甘氨酸单元组成。在pna中,各种嘌呤和嘧啶碱基通过亚甲基羰基键与骨架连接。通常称为不可及rna的锁定核酸(lna)是修饰的rna核苷酸。lna核苷酸的核糖部分用连接2'氧和4'碳的额外桥修饰。该桥将核糖“锁定”在3'-内(北)构象中,这通常在a型双链体中发现。只要需要,lna核苷酸可以与寡核苷酸中的dna或rna残基混合。术语“非结构化核酸”或“una”是含有彼此结合从而降低稳定性的非天然核苷酸的核酸。例如,非结构化核酸可含有g'残基和c'残基,其中这些残基对应于g和c的非天然存在形式,即类似物,它们彼此碱基配对,稳定性降低,但保留分别与天然存在的c和g残基碱基配对的能力。非结构化核酸描述于us20050233340中,其通过引用并入本文以用于una的公开。
如本文所用的术语“寡核苷酸”表示核苷酸的单链多聚体,其长度为约2至200个核苷酸,最多500个核苷酸。寡核苷酸可以是合成的或可以通过酶促制备,并且在一些实施例中,长度为30至150个核苷酸。寡核苷酸可含有核糖核苷酸单体(即,可以是寡核糖核苷酸)和/或脱氧核糖核苷酸单体。例如,寡核苷酸的长度可以是10至20、21至30、31至40、41至50、51至60、61至70、71至80、80至100、100至150或150至200个核苷酸。
“引物”是指天然或合成的寡核苷酸,其在与多核苷酸模板形成双链体时能够用作核酸合成的起始点并且从其3'端沿着模板延伸,使得形成了扩展双链体。在延伸过程中添加的核苷酸序列由模板多核苷酸的序列决定。通常引物通过dna聚合酶延伸。引物通常具有与其在引物延伸产物的合成中的用途相容的长度,并且通常长度为8至100个核苷酸,例如10至75、15至60、15至40、18至30、20至40、21至50、22至45、25至40等,更典型地长度为18至40、20至35、21至30个核苷酸,以及所述范围之间的任何长度。典型的引物的长度可以为10至50个核苷酸,例如15至45、18至40、20至30、21至25等,以及所述范围之间的任何长度。在一些实施例中,引物的长度通常不超过约10、12、15、20、21、22、23、24、25、26、27、28、29、30、35、40、45、50、55、60、65或70个核苷酸。因此,“引物”与模板互补,并通过氢键结合或与模板杂交形成复合物,以产生用于通过聚合酶引发合成的引物/模板复合物,其通过在dna合成过程中添加在其3'端连接的与模板互补的共价键合的碱基而延伸。
术语“杂交(hybridization)”或“杂交(hybridizes)”是指核酸链在正常杂交条件下与第二互补核酸链退火并形成稳定双链体(同源双链体或异源双链体),并且在相同的正常杂交条件下,不与不相关的核酸分子形成稳定双链体。通过在杂交反应中使两条互补核酸链退火来完成双链体的形成。通过调节发生杂交反应的杂交条件(通常称为杂交严格性),可以使杂交反应具有高度特异性,使得两条核酸链之间的杂交不会形成稳定的双链体,例如,在正常严格条件下保留双链区域的双链体,除非两条核酸链在特定序列中含有基本上或完全互补的一定数量的核苷酸。对于任何给定的杂交反应,容易确定“正常杂交或正常严格条件”。参见例如,ausubel等人,《现代分子生物学实验指南(currentprotocolsinmolecularbiology)》,johnwiley&sons,inc.,newyork,或sambrook等人,《分子克隆:实验室手册》,冷泉港实验室出版社(molecularcloning:alaboratorymanual,coldspringharborlaboratorypress)。如本文所用,术语“杂交(hybridizing)”或“杂交(hybridization)”是指核酸链通过碱基配对与互补链结合的任何过程。
如果两个序列在中度至高度严格杂交和洗涤条件下彼此特异性杂交,则认为核酸与参考核酸序列“可选择性杂交”。中度和高度严格杂交条件是已知的(参见例如,ausubel等人,《精编分子生物学实验指南(shortprotocolsinmolecularbiology)》,第3版,wiley&sons1995和sambrook等人,《分子克隆:实验室手册》(molecularcloning:alaboratorymanual),第3版,2001纽约冷泉港实验室出版社)。高度严格条件的一个实例包括在约42℃下在50%甲酰胺、5xssc、5xdenhardt溶液、0.5%sds和100ug/ml变性载体dna中杂交,然后在室温下在2xssc和0.5%sds中洗涤两次,并且在0.1×ssc和0.5%sds中在42℃下再洗涤两次。
如本文所用,术语“双链体”或“双链体化”描述了两个碱基配对的互补多核苷酸,即杂交在一起。
如本文所用的术语“扩增”是指合成与模板核酸的一条链或两条链互补的核酸分子的过程。扩增核酸分子可包括使模板核酸变性,在低于引物的解链温度的温度下使引物与模板核酸退火,并通过酶促从引物延长以产生扩增产物。变性,退火和延长步骤各自可进行一次或多次。在某些情况下,变性,退火和延长步骤进行多次,使得扩增产物的量通常以指数方式增加,尽管本方法不需要指数扩增。扩增通常需要存在脱氧核糖核苷三磷酸,dna聚合酶和适当的缓冲液和/或辅因子以获得聚合酶的最佳活性。术语“扩增产物”是指核酸序列,其由本文定义的扩增方法产生。
术语“确定”、“测量”、“评价”、“评估”、“测定”和“分析”在本文中可互换使用,以指代任何形式的测量,并且包括确定元素是否存在。这些术语包括定量和/或定性测定。评估可以是相对的或绝对的。“评估存在”包括确定存在物的量,以及确定其是否存在。
术语“使用”具有其常规含义,并且因此是指采用例如投入使用的方法或组合物来达到目的。例如,如果使用程序来创建文件,则执行程序以生成文件,该文件通常是程序的输出。在另一个实例中,如果使用计算机文件,则通常访问,读取,并且使用存储在文件中的信息来达到目的。类似地,如果使用唯一标识符,例如条形码,则通常读取唯一标识符以识别例如与唯一标识符相关联的对象或文件。
如本文所用,术语“基因组区域”是指基因组的区域,例如动物或植物基因组,例如人、猴、大鼠、鱼或昆虫或植物的基因组。
如本文所用,术语“基因组序列”是指在基因组中发生的序列。
如本文所用,术语“基因组片段”是指基因组的区域,例如动物或植物基因组,例如人、猴、大鼠、鱼或昆虫或植物的基因组。基因组片段可以是整个染色体,或染色体的片段。基因组片段可以是衔接子连接的(在这种情况下,其具有与片段的一端或两端,或至少与分子的5'端连接的衔接子),或者可以不是衔接子连接的。
如本文所用,术语“连接”是指第一dna分子的5'端的末端核苷酸与第二dna分子的3'端的末端核苷酸的酶催化接合。
“多个”包含至少2个成员。在某些情况下,多个可具有至少2个、至少5个、至少10个、至少100个、至少100个、至少10,000个、至少100,000个、至少106个、至少107个、至少108个或至少109个或更多个成员。
如果两个核酸是“互补的”,则他们在高严格条件下相互杂交。术语“完全互补”用于描述双链体,其中一个核酸的每个碱基与另一个核酸中的互补核苷酸碱基配对。在许多情况下,互补的两个序列具有至少10个,例如至少12或15个具有互补性的核苷酸。
如本文所用的术语“链”是指由通过共价键共价连接在一起的核苷酸构成的核酸,例如磷酸二酯键。在细胞中,dna通常以双链形式存在,因此,具有两条互补的核酸链,在本文中称为“顶部”和“底部”链。在某些情况下,染色体区域的互补链可以称为“正”和“负”链、“第一”和“第二”链、“编码”和“非编码”链、“watson”和“crick”链或“有义”和“反义”链。将链分配为顶部或底部链是任意的,并不暗示任何特定的取向、功能或结构。几个示例性哺乳动物染色体区域(例如,bac、组体、染色体等)的第一链的核苷酸序列是已知的,并且可以在例如ncbi的genbank数据库中找到。
如本文所用,术语“测序”是指通过其获得多核苷酸的至少10个连续核苷酸的身份(例如,至少20个、至少50个、至少100个或至少200个或更多个连续核苷酸的身份)的方法。
术语“新一代测序”是指依诺米那(illumina)、生命科技(lifetechnologies)、太平洋生物科学(pacificbiosciences)和罗氏(roche)等目前采用的所谓的平行化合成测序或连接测序平台。下一代测序方法也可包括纳米孔测序方法或基于电子检测的方法,例如由生命科技商业化的离子激流(iontorrent)技术。
如本文所用,术语“延伸”是指通过添加核苷酸使用聚合酶来延伸引物。如果延伸与核酸退火的引物,则核酸作为延伸反应的模板。
如本文所用,术语“条形码序列”、“分子条形码”或“索引”是指用于(a)鉴定和/或追踪反应中多核苷酸来源和/或(b)计数初始分子测序的次数(例如,在样品中基本上每个分子用不同序列标记,然后扩增样品的情况下)的独特核苷酸序列。条形码序列可以位于寡核苷酸的5'端,3'端或中间,或5'端和3'端。条形码序列的大小和组成可能有很大变化;以下参考文献提供了用于选择适合于特定实施例的条形码序列组的指导:brenner,第5,635,400号美国专利;brenner等人,proc.natl.acad.sci.,97:1665-1670(2000);shoemaker等人,naturegenetics,14:450-456(1996);morris等人,欧洲专利出版物0799897a1;wallace,第5,981,179号美国专利等。在特定实施例中,条形码序列的长度可以为4至36个核苷酸,或6至30个核苷酸,或8至20个核苷酸。
如本文所用,术语“pcr试剂”是指在模板上进行聚合酶链式反应(pcr)所需的所有试剂。如本领域所知,pcr试剂基本上包括第一引物、第二引物、热稳定聚合酶和核苷酸。取决于所用的聚合酶,也可以存在离子(例如mg2+)。pcr试剂可任选地含有可从其扩增靶序列的模板。
术语“相邻”是指小于核苷酸的最长维度的距离。术语“相邻连接”是指两个核苷酸在链上彼此紧邻,并没有插入核苷酸。
在有尾引物或具有5'尾的引物的上下文中,术语“有尾”是指在其5'端具有区域(例如,至少12-50个核苷酸的区域)的引物,该5'端不与引物的3'端杂交到相同的靶标。
术语“可区分序列”是指彼此不同的序列。
如本文所用的术语“靶核酸”是指研究中的目标多核苷酸。
术语“靶核酸分子”是指与其它靶核酸分子一起或不一起存在于组合物中的单个分子。分离的靶核酸分子是指存在于不含其它靶核酸分子的组合物中的单个分子。
在两个或更多个可变核酸序列的上下文中,术语“可变”是指两个或更多个相对于彼此具有不同核苷酸序列的核酸。换句话说,如果群体的多核苷酸具有可变序列,则群体的多核苷酸分子的核苷酸序列在不同分子之间变化。术语“可变”不应理解为要求群体中的每个分子具有与群体中的其它分子不同的序列。
术语“衔接子”是指可以通过连接酶介导的反应与双链dna分子的至少一条链接合的核酸。在一个实施例中,衔接子可以是y衔接子。显而易见的是,衔接子的一端可以被设计成与通过内切核酸酶切割产生的突出端相容,例如,其可以具有平末端或5't突出端。术语“衔接子”是指至少部分双链的分子。衔接子的长度可以是40至150个碱基,例如50至120个碱基,尽管可以设想在该范围之外的衔接子。
术语“y-衔接子”是指包含以下的衔接子:双链区和单链区,其中相对序列是不互补的。双链区的末端可以与靶分子例如基因组dna的双链片段接合,例如通过连接。已经连接到y衔接子的衔接子标记的双链dna的每条链被不对称标记,因为其在一端具有y-衔接子的一条链的序列而在另一端具有y-衔接子的另一条链的序列。在两端已经与y-衔接子接合的核酸分子的扩增产生不对称标记的核酸,即具有含有一个标签序列的5'端和具有另一个标签序列的3'端的核酸。
术语“通用衔接子”是指与研究中的核酸分子的两端连接的衔接子。在某些实施例中,通用衔接子可以是y-衔接子。在两端已经连接到y-衔接子的核酸分子的扩增产生不对称标记的核酸,即具有含有一个标签序列的5'端和具有另一个标签序列的3'端的核酸。
如本文所用,术语“衔接子标记的”是指已被衔接子标记的核酸。衔接子可以接合到核酸分子的5'端和/或3'端。
如本文所用的术语“标记的dna”是指具有添加的衔接子序列的dna分子,即合成来源的“标签”。可以通过连接添加(即“附加”)衔接子序列。
如本文所用,术语“单独消化”是指两种或更多种不同的切割反应。
如本文所用,术语“核酸指导的内切核酸酶”是指dna和rna指导的内切核酸酶,其包括argonaut和ii型crispr/基于cas的系统,该系统由两种组分组成:切割靶dna的核酸酶(例如,cas9内切核酸酶或其变体或直向同源物)和将核酸酶靶向靶dna中的特定位点的指导rna(grna)。参见,例如,hsu等人(naturebiotechnology201331:827–832)。
如本文所用,术语“部分”是指样品的一部分(例如,等分试样)。
如本文所用,术语“确定的位点”是指选定的序列。
如本文所用,术语“选择性扩增”是指仅选择的序列被扩增的扩增反应(例如,pcr反应)。
在某些情况下,本文所述方法中使用的寡核苷酸可以使用参考基因组区域设计,即具有已知核苷酸序列的基因组区域,例如其序列例如保存在ncbi的genbank数据库或其它数据库中的染色体区域。这种寡核苷酸可用于使用含有测试基因组的样品的测定中,其中测试基因组含有寡核苷酸的结合位点。
如本文所用,术语“生物素部分”是指包括包括生物素或生物素类似物的亲和剂,例如脱硫生物素、氧代生物素、2-亚氨基生物素、二氨基生物素、生物素亚砜、生物胞素等。生物素部分与链霉抗生物素蛋白结合,亲和力为至少10-8m。生物素亲和剂还可以包括接头,例如-lc-生物素、-lc-lc-生物素、-slc-生物素或-pegn-生物素,其中n是3-12。
术语“亲和标签”和“捕获部分”是指能够:a)非共价特异性地彼此结合或b)选择性地彼此反应以形成共价键的部分。非共价特异性地彼此结合的合适亲和标签和捕获剂对的实例有很多,包括但不限于:链霉抗生物素蛋白/抗生物素蛋白、洋地黄毒苷/抗洋地黄毒苷抗体、荧光素/抗荧光素抗体,尽管许多其它是已知的。选择性地彼此反应以形成共价键的化学选择性反应性基团的实例有很多,并包括:胺和活性酯例如nhs酯、硫醇和马来酰亚胺或碘乙酰胺),以及可通过click化学彼此反应的基团,例如叠氮化物和炔烃基团。含有可用于本文的亲和标签的核糖核苷酸可从许多来源商购获得。
术语“末端封端”是指已被修饰以使其不能用于连接的末端。尽管可以使用其它方法,但可以通过用磷酸酶处理来末端封端核酸。
具体实施方式
在一些实施例中,该方法可以包括使用多种重编程的核酸指导的内切核酸酶消化混合核酸样品(即,含有来自至少两个来源的dna的核酸样品,其中来自一个来源的dna可以占样品中总dna的小于10%、小于5%、小于2%或小于1%),所述内切核酸酶靶向目标序列(即一组cas9内切核酸酶,argonaut,其直向同源物或变体,其具有将内切核酸酶靶向靶核酸(例如靶基因组)中的预定靶位点的合成指导rna或dna)。该步骤产生在样品中包含核酸片段的消化样品,其中消化样品中的至少一些片段包含:(i)目标序列和(ii)至少一个通过内切核酸酶切割产生的可连接末端(即,含有5'磷酸和3'羟基的末端,其末端是平的或具有限定的突出端)。一些核酸指导的内切核酸酶(包括cas9)产生平末端,而其它可产生限定的突出端。消化后,可以富集含有目的序列的片段,例如,使用大小选择,通过将包含捕获部分(例如,生物素部分)的衔接子连接到由内切核酸酶产生的可连接末端,将捕获部分结合到载体(例如,链霉抗生物素蛋白载体),洗去未结合的核酸,或通过将衔接子(例如“y-衔接子”)连接到片段的末端,并使用与y衔接子中的序列结合或互补的引物通过pcr扩增连接的片段。可以通过任何合适的方法分析富集的片段,例如测序。
对于cas9,指导rna可以由两个分子组成,即一个与靶杂交并提供序列特异性的rna(“crrna”)和一个能够与crrna杂交的rna(“tracrrna”)。或者,指导rna可以是含有crrna和tracrrna序列的单个分子(即sgrna)。cas9蛋白与野生型cas9蛋白(例如,化脓性链球菌(streptococcuspyogenes)cas9蛋白)具有至少60%同一性(例如,至少70%、至少80%、或90%同一性、至少95%同一性或至少98%同一性或至少99%同一性)。cas9蛋白可具有野生型cas9蛋白的所有功能,或仅具有一种或一些功能,包括结合活性和核酸酶活性。
为了使cas9成功结合dna,基因组dna中的靶序列应该与grna序列互补,并且必须紧接着是正确的原型间隔区相邻基序或“pam”序列。pam序列存在于dna靶序列中但不存在于grna序列中。其后是pam序列的具有正确靶序列的任何dna序列将被cas9结合。pam序列根据产生cas9的细菌物种而不同。最广泛使用的ii型crispr系统源自化脓性链球菌,pam序列是位于grna识别序列的紧邻3'端的ngg。来自示例性细菌物种的ii型crispr系统的pam序列包括:化脓性链球菌(ngg)、脑膜炎奈瑟氏球菌(neisseriameningitidis)(nnnngatt)、嗜热链球菌(streptococcusthermophilus)(nnagaa)和齿垢密螺旋体(treponemadenticola)(naaaac)。对于一些其它序列特异性核酸酶,例如argonauts,不需要pam位点来结合和切割靶dna。
显而易见的是,该反应可以在体外进行,即在无细胞环境中使用分离的核酸(例如分离的dna)进行。混合样品可以从任何来源收集,包括任何生物、有机材料或含核酸的物质,包括但不限于植物、动物(例如爬行动物、哺乳动物、昆虫、蠕虫、鱼等)、组织样品、细菌、真菌(例如酵母)、噬菌体、病毒、尸体组织、考古/古代样品等。在某些实施例中,该方法中使用的基因组dna可以源自哺乳动物,其中在某些实施例中,哺乳动物是人类。
例如,该方法中使用的指导rna可以经设计使得他们指导内切核酸酶与靶基因组中的预定切割位点的结合。在某些情况下,可以选择切割位点以释放含有未知序列区域或含有snp、核苷酸插入、核苷酸缺失、重排等的区域的片段。由于许多生物的基因组分离方法和核苷酸序列(包括许多细菌、真菌、植物和动物,例如哺乳动物,例如人、灵长类动物和啮齿动物,例如小鼠和大鼠)是已知的,因此设计用于本方法的指导rna应该在本领域技术人员的技能范围内。例如,cas9-grna复合物可以被编程为与任何序列结合,条件是该序列具有pam基序。理论上,cas9-grna复合物可切割基因组dna以产生30-50bp的片段。然而,实际上,切割位点之间的最小间隔可以例如在50-900bp的范围内。在一些实施例中,sgrna或crrna可以是简并序列以靶向相对保守的区域。
该方法可以使用一组至少2种、至少5种、至少10种、至少100种、至少1,000种、至少10,000种、至少50,000种或至少100,000种或更多种不同的指导rna/dna,其各自与一个或多个基因组中不同的,预定义的位点互补。相邻位点之间的距离可以根据所需的应用而极大地变化。在一些实施例中,相邻位点之间的距离可以在100bp至200kb的范围内,并且在特定实施例中,可以选择位点以释放在限定的大小范围内的片段,例如100bp至2kb,例如,200bp至1kb,或更大(例如,500bp至20kb)用于纳米孔和/或pacbio测序。在某些情况下,可以选择指导rna/dna以释放具有适合于大小选择的大小(例如,小于1kb或小于500bp)的片段。在这些实施例中,片段的长度可小于1kb或长度大于约1kb,并且可通过选择片段的大小来完成富集。
在一些实施例中,内切核酸酶的消化可以产生具有两个末端的片段,两个末端都是可连接的。在这些实施例中,该方法包括将衔接子连接到片段的两端,从而允许使用与衔接子或其补体杂交的单对引物通过pcr富集连接的片段。在这些实施例中,片段还可以在他们连接到衔接子之前或之后通过大小富集。显而易见的是,所用的衔接子应与内切核酸酶产生的末端相容。在一些实施例中,与片段连接的衔接子的末端可以是平端的。在其它实施例中,与片段连接的衔接子的末端可以具有与内切核酸酶产生的突出端互补的突出端。在进一步的实施例中,在连接到t尾衔接子之前,平末端的片段可以是a尾(例如,使用taq聚合酶)。如上所述,在一些实施例中,衔接子可以是y-衔接子,并且因此,连接产物的每条链可以被不对称地标记,因为其在一端具有y-衔接子的一条链的序列,并且在另一端具有y-衔接子的另一条链的序列,其中添加的序列不相同或互补。在两端已经与y-衔接子接合的核酸分子的扩增产生不对称标记的核酸,即具有含有一个标签序列的5'端和具有另一个标签序列的3'端的核酸。
在一些实施例中,衔接子可含有索引序列,例如,在分子之间变化的序列,例如随机序列,从而允许对分子进行计数。在这些实施例中,该方法可包括将编索引的衔接子连接到片段的两端,扩增连接的片段,对连接的片段测序以产生序列读段,然后对与序列读段中的目标序列相联的分子索引器序列的数量进行计数,从而提供核酸样品中目标序列的拷贝数的估计值。这允许用户评估原始样品中不同序列分子的相对量。
该方法可用于获得重叠序列读段,从而允许重叠群的组装。在这些实施例中,该方法可以包括:(a)单独(例如,在单独的容器中):(i)使用靶向目标序列的第一多种重编程的核酸指导的内切核酸酶消化混合核酸样品的第一部分,以产生第一消化样品,其中消化样品中的至少一些片段包含:(i)第一目标序列和(ii)至少一个通过内切核酸酶切割产生的可连接末端;(ii)使用靶向目标序列的第二多种重编程的核酸指导的内切核酸酶消化混合核酸样品的第二部分,以产生第二消化样品,其中消化样品中的至少一些片段包含:(i)第二目标序列和(ii)至少一个通过内切核酸酶切割产生的可连接末端。在该方法中,可以设计每次消化中的内切核酸酶,使得第一消化样品中的至少一些片段与第二消化样品中的至少一些片段重叠,例如,重叠至少50、至少100或至少200个碱基)。可以如上所述富集含有目标序列的片段(例如,通过连接衔接子并使用与衔接子中的序列或其补体杂交的引物通过pcr扩增片段),然后测序以产生多个序列读段。在测序之后,该方法可以包括组装任何重叠序列读段,从而获得包含至少部分第一目标序列和至少部分第二目标序列的序列的重叠群。
显而易见的是,用于扩增的衔接子和/或引物可以与在其中使用引物延伸的任何下一代测序平台中的使用相容,例如,依诺米那的可逆终止子方法、罗氏的焦磷酸测序方法(454)、生命科技的通过连接的测序(solid平台)、生命科技的iontorrent平台或太平洋生物科学'荧光碱基切割方法。这些方法的实例描述于以下参考文献中:margulies等人(nature2005437:376–80);ronaghi等人(analyticalbiochemistry1996242:84–9);shendure(science2005309:1728);imelfort等人(briefbioinform.200910:609-18);fox等人(methodsmolbiol.2009;553:79-108);appleby等人(methodsmolbiol.2009;513:19-39)english(plosone.20127:e47768)和morozova(genomics.200892:255-64),为了方法的和方法的特定步骤的一般描述,包括每个步骤的所有起始产物,试剂和最终产物,将他们通过引用并入。实际上,如果产物在固体载体上扩增(例如,使用依诺米那流动池),那么扩增子可以在基质上就地测序。测序步骤可以使用任何方便的下一代测序方法完成,并且可以产生至少10,000、至少50,000、至少100,000、至少500,000、至少1m、至少10m、至少100m或至少1b个序列读段。在许多情况下,读数是双端读数。
在任何实施例中,在消化之前,该方法可以包括对混合核酸样品进行末端封端,使得核酸片段的末端不可用于连接。例如,在上述任何实施例中,在消化之前,该方法可以包括用磷酸酶(例如,cip、sap、plap或seap)处理混合核酸样品,从而从样品的核酸中除去5'磷酸基团并使样品中核酸的5'端不可连接。替代的封端方法也是可能的,包括发夹衔接子的连接,含有化学阻断基团的衔接子的连接,缺乏5'磷酸的衔接子的连接,封端基团的化学添加,修饰核苷酸的酶介导的添加,酶介导的一个或多个核苷酸的添加,产生粘性末端突出端,其与特定衔接子的未来连接不相容,或任何其它阻止功能性衔接子的有效下游连接的方法。
在一些实施例中,所分析的核酸可以源自单一来源(例如,来自不同位点或单个受试者中的时间过程),而在其它实施例中,核酸样品可以是从多个不同来源中提取的核酸文库(例如,来自不同受试者的核酸文库),其中“多个”是指两个或更多个。因此,在某些实施例中,核酸样品可含有来自2个或更多个来源、3个或更多个来源、5个或更多个来源、10个或更多个来源、50个或更多个来源、100个或更多个来源、500个或更多个来源、1000个或更多个来源、5000个或更多个来源、最多并包括约10,000个或更多个来源的核酸。分子条形码可以允许来自不同来源的序列在分析后进行区分。
在一些实施例中,dna样品可以与封端的衔接子连接以防止扩增。在扩增之前,可以用靶向切口酶(例如cas9d10a)处理连接的样品。切口产生将允许靶向序列的延伸和仅在与这些序列附接的衔接子中置换封端基团,允许切口分子的pcr扩增,而不能扩增非靶向分子。封端基团的实例包括但不限于脱氧尿嘧啶(du)、反向脱氧胸苷(dt)、rna碱基、脱氧异胞啶(isodc)、脱氧异鸟嘌呤(isodg)、1',2'-双脱氧核苷酸(ddntp)、间隔基、无碱基位点等。该方法的实施例在图4中示出。该方法可包括将衔接子连接至片段的末端,其中衔接子含有封端基团(即,导致聚合酶停滞的基团),使用核酸指导的切口内切核酸酶(例如,cas9d10a)切割靶片段,使带切口的链延伸以置换含有封端基团的链,然后使用与衔接子杂交的通用引物扩增靶序列。
在一些实施例中,核酸样品可包含来自至少两种生物的dna,例如哺乳动物和病原体(例如,其中病原体可以是病毒、细菌或真菌),哺乳动物和微生物菌群,或微生物菌群中的两种微生物,其中来自一种生物的dna的丰度可以是另一种的至少10倍、至少50倍、或至少100倍、至少500倍或至少1,000倍。在一些实施例中,核酸样品可含有来自相同生物(例如癌症患者)的野生型和突变体dna。在一些实施例中,混合样品可以是环境样品、来自犯罪现场的样品或考古样品。在一些实施例中,混合样品由临床样品制成,例如来自疑似已被病原体感染的患者。临床样品可以是下面列出的体液或排泄物。在一些实施例中,临床样品可以是肿瘤活检样品。从各种样品(例如临床、法医和环境样品)中提取总dna和rna的方法是本领域熟知的。样品包括但不限于皮肤拭子、皮肤活检、唾液、牙拭子、牙齿刮片、面颊拭子、咽拭子、痰、内脏样本、粪便、尿液、阴道、宫颈、宫颈内膜、子宫内膜、鼻拭子、肺、器官活检和组织活检。样品也可以是体液。感兴趣的体液包括但不限于羊水、房水、玻璃体液、血液(例如全血、分馏血液、血浆、血清等)、母乳、脑脊液(csf)、耳垢(耳垢)、乳糜、边缘、内淋巴、外淋巴、粪便、胃酸、胃液、淋巴液、粘液(包括鼻腔引流和痰)、心包液、腹膜液、胸膜液、脓液、大黄、唾液、皮脂(皮油)、精液、痰液、汗液、滑液、泪液、呕吐物和尿液。在特定实施例中,样品可以是临床样品,例如从患者收集的样品。在某些情况下,这些样品中的dna可以高度片段化成,例如平均大小为10bp至5kb,例如20bp至200bp,并且在某些情况下可以使用本文所述的方法进行片段化。从这些样品中提取总dna的方法是众所周知的。
上述方法可用于分析各种诊断、药物发现和研究应用中的样品。上述方法可用于分析生物样品。如本文所用,术语“生物样品”是指从生物或从生物的组分(例如细胞)获得的样品。样品可以是任何生物组织或流体。在一些情况下,样品将是“临床样品”,其是源自患者的样品。这些样品包括但不限于痰液、血液、血细胞(例如白细胞)、组织或细针活检样品、尿液、腹膜液和胸膜液、或来自其的细胞。生物样品还可包括组织切片,例如用于组织学目的的冷冻切片。本方法还可用于确定水、污水、空气样品、食品,包括动物、蔬菜、种子等、土壤样品、植物样品、微生物培养样品、细胞培养样品、组织培养样品中微生物的特性,以及用于人类医学、兽医学、农业、食品科学、生物威胁和工业微生物学等。本方法允许因不需要培养微生物而鉴定难以培养微生物。因此,本方法提供了对样品中微生物的快速检测,不需要等待培养微生物的时间。
在一些实施例中,该方法可用于从临床样品中鉴定微生物病原体。在这些实施例中,内切核酸酶可以靶向来自多种不同病原体(例如,至少10种或至少100种不同病原体)的序列,在不知道哪种病原体负责感染下,可以对富集的核酸进行测序,并且可以将序列与来自已知病原体(例如细菌、东方和病毒病原体)的序列相比较,并且如果发现匹配,则可以将受试者诊断为被该病原体感染。在一些实施例中,核酸酶可以用简并识别序列编程,所述简并识别序列代表编码目标分子途径中涉及的一个或多个特定基因的已知保守基序。以这种方式,可以同时对来自几个物种的同源基因富集和测序。在另一个实施例中,该方法可用于宿主相关群落(“微生物菌群”)的深度测序,其优点在于该方法能够对样品中的所有微生物dna进行测序,而不仅仅是dna的子集,例如,仅核糖体dna。可以使用本方法,组合物和试剂盒鉴定的微生物包括但不限于:多种革兰氏(+)细菌、多种革兰氏(-)细菌、肠杆菌科(enterobacteriaceae)中的多种细菌、肠球菌属(enterococcus)中的多种细菌、葡萄球菌属(staphylococcus)中的多种细菌、以及弯曲杆菌属(campylobacter)、大肠杆菌(e.coli)、各种菌株的大肠杆菌例如,k12-mg1655、cft073、o157:h7edl933、o157:h7vt2-sakai等、肺炎链球菌(streptococcuspneumoniae)、铜绿假单胞菌(pseudomonasaeruginosa)、金黄色葡萄球菌(staphylococcusaureus)、凝固酶阴性葡萄球菌、多种念珠菌属(candida),包括白色念珠菌(c.albicans)、热带念珠菌(c.tropicalis)、都柏林念珠菌(c.dubliniensis)、维斯念珠菌(c.viswanathii)、近平滑念珠菌(c.parapsilosis)、克雷白氏杆菌(klebsiellapneumoniae)、多种分枝杆菌属(mycobacterium),例如结核分枝杆菌(m.tuberculosis)、牛分支杆菌(m.bovis)、牛分支杆菌bcg、瘰疬分枝杆菌(m.scrofulaceum)、堪萨斯分枝杆菌(m.kansasii)、龟分支杆菌(m.chelonae)、戈氏分枝杆菌(m.gordonae)、溃疡分枝杆菌(m.ulcerans)、日内瓦分枝杆菌(m.genavense)、m.xenoi、猿分枝杆菌(m.simiae)、偶发分枝杆菌(m.fortuitum)、海鱼分枝杆菌(m.malmoense)、隐藏分枝杆菌(m.celatum)、嗜血分支杆菌(m.haemophilumand)和非洲分枝杆菌(m.africanum)、利斯特氏菌属(listeria)、布鲁氏菌属(brucella)、耶尔森氏菌属(yersinia)等中的多种细菌。因此,本方法能够将微生物鉴定到微生物的属、种、亚种、菌株或变体的水平。
疾病状态可能表现出新微生物的存在,正常微生物的不存在,或微生物比例的改变。疾病状态也可具有与正常状态基本相似的微生物群体,但由于环境或宿主遗传因素而具有不同的微生物功能或对微生物的不同宿主反应。例如,最近的研究已经确定,宿主与其微生物群之间的正常平衡的破坏(通常表现为微生物失衡)与许多病症和疾病相关并且可能导致许多病症和疾病。这些尤其包括克罗恩病、溃疡性结肠炎、肥胖症、哮喘、过敏症、代谢综合征、糖尿病、牛皮癣、湿疹、酒渣鼻、特应性皮炎、胃肠道反流病、胃肠道癌症、细菌性阴道病、神经发育性疾病如自闭症谱系障碍、以及多种感染。例如,在克罗恩病中,拟杆菌、真细菌和蛋白胨链球菌属的浓度增加,而双歧杆菌数量减少(linskens等人,scandjgastroenterolsuppl.2001;(234):29-40);在溃疡性结肠炎中,兼性厌氧菌的数量增加。在这些炎性肠病中,这种微生物失衡导致增加的免疫刺激和增强的粘膜通透性(sartor,procnatlacadsciusa.2008年10月28日;105(43):16413-4)。在肥胖受试者中,相对于瘦人而言,拟杆菌的相对比例已显示出降低(ley等人,nature.2006年12月21日;444(7122):1022-3),以及也已经讨论了微生物失衡与糖尿病发展的可能联系(cani等人,patholbiol(巴黎).2008年7月;56(5):305-9)。在皮肤中,已经在传染性和非传染性疾病和障碍中提出了土着微生物群在健康和疾病中的作用,例如特应性皮炎、湿疹、红斑痤疮、牛皮癣和痤疮(holland等人br.j.dermatol.96:623-626;thomsen等人arch.dermatol.116:1031-1034;till等人br.j.dermatol.142:885-892;paulino等人j.clin.microbiol.44:2933-2941)。此外,常驻微生物群也可能因皮肤屏障受损而致病(roth和jamesannurevmicrobiol。1988;42:441-64)。细菌性阴道病是由天然存在的阴道微生物群的失衡引起的。虽然正常的阴道微生物群以乳杆菌为主,但在2级(中间)细菌性阴道病中,除乳杆菌外,还存在加德纳菌(gardnerella)和动物溃疡伯格菌(mobiluncusspp.)。在3级(细菌性阴道病)中,加德纳菌和动物溃疡伯格菌、以及乳杆菌很少或不存在(hay等人,br.med.j.,308,295-298,1994)。鉴定哪些微生物在样品中可以帮助诊断和治疗这些疾病。
此外,该方法可用于检测rna病毒或逆转录病毒,例如呼肠孤病毒、轮状病毒、肠病毒、鼻病毒、肝病毒、心脏病毒、口疮病毒、脊髓灰质炎病毒、双埃柯病毒、马鼻病毒、嵴病毒、捷申病毒、柯萨奇病毒、诺瓦克病毒、风疹病毒、甲病毒、淋巴细胞性脉络丛脑膜炎病毒、登革热病毒、丙型肝炎病毒、黄热病毒、甲型流感病毒、乙型流感病毒、丙型流感病毒、贫病毒、首戈托病毒、麻疹病毒、腮腺炎病毒、呼吸道合胞病毒、牛瘟病毒、犬瘟热病毒、加利福尼亚脑炎病毒、汉坦病毒、狂犬病病毒、埃博拉病毒、马尔堡病毒、冠状病毒、星状病毒、博纳病病毒、动脉病毒、马动脉炎病毒、戊型肝炎病毒、逆转录病毒(例如,hiv-1和hiv-2)以及乙型肝炎病毒。
该方法还可用于检测微生物dna或rna序列并同时检测人dna或rna序列,从而允许在同一实验中研究病原体和宿主。
该方法也可用于基因分型。在这种情况下,其将靶向病原体或其它物种内的特定位点,这些位点可用于区分特定菌株或其它亚群。在感染了多个亚群或检测微生物菌群或环境样品中的多个亚群的情况下,施用或未施用唯一分子标识符(umi)的该方法可用于评价不同亚群的相对量。
在一些实施例中,该方法可用于确定核酸酶的靶序列,例如限制性内切核酸酶、归巢内切核酸酶和可编程核酸酶,例如cas9或cpf1。例如,为了鉴定特定cas9指导rna组合的切割位点,可以用cas9和指导rna处理用封端的衔接子合成的基因组dna文库,以允许选择性扩增含有切割位点的序列。
该方法的这些和其它用途将是显而易见的。
试剂盒
本公开还提供了用于实施如上所述的本方法的试剂盒。在某些实施例中,本试剂盒可以包含:a)多种重编程的核酸指导的内切核酸酶;以及b)磷酸酶或其它封端试剂。这样的试剂盒可任选地含有衔接子和pcr引物,其中pcr引物的3'端具有与衔接子中的序列相同或互补的序列。以上描述了该试剂盒的组分的进一步细节。试剂盒还可以含有上文和下文所述的可以在该方法中使用的其它试剂,例如连接酶、聚合酶等,这取决于该方法将如何实施。
除了上述组分之外,本试剂盒还包括使用试剂盒的组分来实施本方法的说明书。用于实施本方法的说明书通常记录在合适的记录介质上。例如,说明书可以印刷在基材上,例如纸或塑料等。因此,说明书可以作为包装插页存在于试剂盒中,存在于试剂盒的容器或其组件的标签中(即与包装或分包装相关联)等。在其它实施例中,说明书作为存在于合适的计算机可读存储介质上的电子存储数据文件存在,例如cd-rom、软盘等。在又其它实施例中,实际说明书不存在于试剂盒中,而是提供用于例如通过互联网从远程源获得说明书的手段。该实施例的实例是包括网址的试剂盒,在该网址中可以查看说明书和/或可以从该网址下载说明书。与说明书一样,这种用于获得说明书的手段记录在合适的基材上。
为了进一步说明本发明,给出以下具体实施例,应理解,提供他们是为了说明本发明,并且不应以任何方式解释为限制其范围。
实施例
实施例1a.一种样品分析方法,其包括:(a)使用多种重编程的核酸指导的内切核酸酶消化末端封端的混合核酸样品以产生消化样品,所述内切核酸酶靶向目标序列,其中消化样品中至少一些片段包含:(i)目标序列和(ii)至少一个通过内切核酸酶切割产生的可连接末端;(b)富集含有目标序列的片段;以及(c)分析富集的片段。
实施例1b.一种样品分析方法,其包括:
(a)使用多种重编程的核酸指导的内切核酸酶消化磷酸酶处理的混合核酸样品以产生消化样品,所述内切核酸酶靶向目标序列,其中消化样品中至少一些片段包含:(i)目标序列和(ii)至少一个通过内切核酸酶切割产生的可连接末端;(b)富集含有目标序列的片段;以及(c)分析富集的片段。
实施例2.根据实施例1a或b所述的方法,其中所述方法包括将衔接子连接至步骤(a)中通过内切核酸酶切割产生的可连接末端。
实施例3.根据实施例2所述的方法,其中所述衔接子包含捕获部分,并且所述富集通过将捕获部分与载体结合并洗去未结合的核酸来进行。
实施例4.根据以上任一实施例所述的方法,其中(a)的内切核酸酶消化产生在限定大小范围内的片段。
实施例5.根据实施例4所述的方法,其中所述富集通过选择片段的大小来进行。
实施例6.根据以上任一实施例所述的方法,其中(a)的内切核酸酶消化产生两端均可连接的片段。
实施例7.根据以上任一实施例所述的方法,其中(a)的消化产生在两端具有可连接的内切核酸酶切割位点的片段,并且所述方法包括将衔接子连接到片段的两端。
实施例8.根据实施例7所述的方法,其中所述富集通过使用与衔接子或其补体杂交的引物扩增连接的片段来进行。
实施例9.根据实施例8所述的方法,其中所述衔接子含有分子索引器。
实施例10.根据以上任一实施例所述的方法,其中所述方法包括将编索引的衔接子连接到片段的两端,其中所述索引是一系列随机核苷酸,其长度足以使每个片段有较高概率接受不同索引;对连接的片段测序以产生序列读段;然后对与序列读段中的目标序列相联的分子索引器序列的数量进行计数,从而提供核酸样品中目标序列的拷贝数的估计值。
实施例11.根据以上任一实施例所述的方法,其中所述方法包括:
(a)单独地:
(i)使用靶向目标序列的多种重编程的核酸指导的内切核酸酶消化混合核酸样品的第一部分,以产生第一消化样品,其中消化样品中的至少一些片段包含:(i)第一目标序列和(ii)至少一个通过内切核酸酶切割产生的可连接末端;
(ii)使用靶向目标序列的多种重编程的核酸指导的内切核酸酶消化混合核酸样品的第二部分,以产生第二消化样品,其中消化样品中的至少一些片段包含:(i)第二目标序列和(ii)至少一个通过内切核酸酶切割产生的可连接末端;
其中第一消化样品中的至少一些片段与第二消化样品中的至少一些片段重叠;
(b)富集含有目标序列的片段;
(c)对富集的序列进行测序以产生多个序列读段;以及
(d)组装重叠序列读段,从而获得第一目标序列和第二目标序列的重叠群。
实施例12.根据以上任一实施例所述的方法,其中通过内切核酸酶处理产生的可连接末端是平末端。
实施例13.根据以上任一实施例所述的方法,其中混合核酸样品包含来自至少两种生物的dna。
实施例14.根据实施例13所述的方法,其中至少两种生物包含哺乳动物和病原体。
实施例15.根据实施例14所述的方法,其中所述病原体是病毒、细菌或真菌。
实施例16.根据实施例15所述的方法,其中所述至少两种生物包含哺乳动物和微生物菌群。
实施例17.根据以上任一实施例所述的方法,其中所述混合样品包含野生型和突变体dna。
实施例18.根据以上任一实施例所述的方法,其中所述混合样品是环境样品、来自犯罪现场的样品或考古样品。
实施例19.根据以上任一实施例所述的方法,其中所述混合样品由临床样品构成。
实施例20.根据实施例19所述的方法,其中所述临床样品是体液或排泄物。
实施例21.根据实施例20所述的方法,其中所述临床样品是血液、痰或粪便。
实施例22.根据实施例20所述的方法,其中所述临床样品是肿瘤活检样品。
实施例23.根据以上任一实施例所述的方法,其中(c)中的分析通过测序进行。
实施例24.根据以上任一实施例所述的方法,其中所述内切核酸酶是cas9或argonaught、其直向同源物或其变体。
实施例25.根据以上任一实施例所述的方法,其中所述样品通过至少2种重编程的核酸指导的内切核酸酶消化。
实施例26.根据以上任一实施例所述的方法,其中在步骤(a)之前,所述方法包括用磷酸酶处理混合核酸样品,从而使样品中核酸的5'端不可连接。
实施例27a.一种包含多种重编程的核酸指导的内切核酸酶;和磷酸酶的试剂盒。
实施例27b.一种包含多种重编程的核酸指导的内切核酸酶;和磷酸酶或其它末端封端试剂的试剂盒。
实施例28.根据实施例27a或b所述的试剂盒,还包含衔接子。
实施例29.根据实施例27a或b所述的试剂盒,还包含pcr引物,其中所述pcr引物的3'端具有与衔接子中的序列相同或互补的序列。
实例
给出以下实例是为了说明本发明的各种实施例,并不意味着以任何方式限制本发明。本实例以及本文所述的方法目前代表优选实施例,是示例性的,并不意图作为对本发明范围的限制。本领域技术人员将想到其中的变化和包含在由权利要求的范围限定的本发明的精神内的其它用途。
使用flash靶向抗生素抗性基因,证明相对于源自患者样品的随机片段化cdna文库,如meca的序列的富集超过10倍。
cas9-dgrna复合物的设计与构建
在本研究中选择了13种能够在金黄葡萄球菌中赋予抗生素抗性的基因用于flash。九种(meca、qnr、qacb、mfs、mdea、nora、norb/c、kctc、和pao1)是质粒携带基因,其单独存在指示抗性;另外四种(促旋酶a、促旋酶b、parc和pare)是金黄葡萄球菌内源性基因,已知其特定点突变会引起喹诺酮抗性。对于质粒携带基因,在基因两端附近选择指导rna位点,并且在基因内约每隔200-300bp。对于具有点突变的基因,侧接每个突变选择两个位点,其距离将产生200-300bp的切割产物。指导rna的列表及其在基因内的位置可以在下表1中找到。
表1:
由于降低了成本,因此选择双指导rna而不是单指导rna。对于每个选择的靶标,设计了60mercrrna模板寡核苷酸,其包括18个碱基的t7转录起始位点,靶向20mer和crrna的剩余22个碱基;购买该序列的反向互补(5′-caaaacagcatagctctaaaacnnnnnnnnnnnnnnnnnnnnctatagtgagtcgtatta-3′)(seqidno:64)。所有寡核苷酸均购自idt(integrateddnatechnologies,coralville,ia,usa)。汇集所有crrna用于转录。使用定制的t7rna聚合酶(rnap)进行转录。在每个100μl反应中,将400ngdna模板与t7rnap(最终浓度为8ng/μl),缓冲液(最终浓度为40mmtrisph8.0、20mmmgcl2、5mmdtt和2mm亚精胺)和ntp(thermofisherscientific,waltham,ma,usa)(最终浓度为各1mmatp、ctp、gtp和utp)混合,并在37℃下温育4小时。典型的产量为10至50μgrna。用zymornaclean&concentrator-5试剂盒(zymoresearch,irvine,ca,usa)纯化crrna。
合成tracrrna获自idt。通过以等摩尔浓度混合两种组分,将混合物在95度下加热1分钟,然后在操作台上将其缓慢冷却至室温,将crrna文库与tracrrna复合。将得到的dgrna等分,保存在-80℃,解冻后仅使用一次。
如gu和crawford等人所述,表达并纯化用mruby2标记的cas9。2016[1].
flash和深度测序培养物和患者样品
使用标准程序从患者培养分离物(样品31和33)制备基因组dna。使用用于低核酸含量样品的nugenovationv.2试剂盒(nugen,sancarlos,ca,usa)由患者支气管肺泡灌洗(bal)液(样品288和1002)制备cdna,如[2,3]中所述。将来自每个样品的10纳克dna用小牛肠磷酸酶(cip)在37℃处理30分钟,以使存在的所有5'dna末端去磷酸化。将样品自旋柱纯化以除去cip。通过在1xcas9活性缓冲液(最终浓度为50mmtrisph8.0、100mmnacl、10mmmgcl2和1mmtcep)中将浓度各40μm的rna文库与cas9混合,并在37℃温育10分钟来形成rnp复合物。然后将cas9-指导rna复合物加入到cip处理的dna的1xcas9活性缓冲液中,最终浓度为10umrnp,总体积为20μl。在37℃温育2小时后,用蛋白酶k在37℃下处理样品15分钟,然后在95℃煮沸5分钟。然后用ampure珠以1:1.4的比率清洗样品。将他们在20μl水中洗脱。
作为对照,根据制造商的说明书,用nebnext片段化酶处理另10ng各dna样品25分钟。
根据制造商的说明书,使用nebnextultraii文库制备试剂盒将cas9切割的dna和片段化酶切割的dna样品转化为依诺米那测序文库,并用双索引条形码引物扩增。
用ampure珠以1:0.9的比率纯化扩增样品,然后通过qubit定量并以相等的浓度合并。生物分析仪高灵敏度dna试剂盒迹线在约140bp处显示大峰,表明衔接子二聚体。因此,样品用ampure珠以1:0.8的比率再次纯化,并使用kapa文库扩增试剂盒和依诺米那通用p5和p7引物通过6次以上的pcr循环进行扩增。重复的生物分析仪迹线显示,已经去除了140bp处的峰,并且保留了以200-300bp为中心的宽峰。通过ddpcr(bio-rad,hercules,ca,usa)定量文库。使用125bp双端测序在依诺米那hiseq2500仪器中对两个泳道的部分进行测序。
数据使用priceseqfilter[4]过滤,然后通过内部病原体发现管道(如gu和crawford等人2016[1]所述)。
结果
图3描绘了用flash富集靶向抗生素抗性基因。在没有flash的情况下,meca在培养分离物中很容易检测到,但在由患者样品制备的cdna中难以检测到。flash将meca覆盖率提高了12倍。
讨论
flash方法不限于文中描述的方案。在其它情况下,除了衔接子连接,可以通过大小选择分离释放的dna部分,然后在分离后扩增或连接。在已知特定目标基因中仅一个可切割序列的情况下,序列特异性核酸酶消化后可以连接附着于标记(例如生物素)的化学标记的寡核苷酸,以捕获和纯化所需序列。然后可以使用随机片段化,然后进行另一个衔接子连接步骤来纯化和扩增含有原始序列的dna分子。
为了向该技术添加定量方面,衔接子可以包括umi(唯一分子标识符)序列。umi是与衔接子或索引引物上的条形码相邻的简并序列(例如nnnnnnnnnnnn)。如果置于索引引物上,在索引pcr的第一个循环中,样品中的各单个分子将用含有相同条形码但不同的唯一umi的引物扩增。在随后的循环中,将扩增umi序列。当对文库进行测序时,对每个测序片段计数的umi数量表明核酸酶处理后原始样品中存在的该片段的拷贝数。
此外,可以用简并序列构建指导rna,以靶向可能含有未知物种(或其基因组未测序的已知物种)的样品中的序列的保守区域。例如,这将允许剖析如人类微生物菌群样品的宏基因组样品中存在的代谢能力。
参考文献
1.guw,crawforded,o'donovanbd,wilsonmr,chowed,retallackh等人,通过杂交耗竭高丰度序列(dash):在测序文库和分子计数应用中使用cas9去除不想要的高丰度物种(depletionofabundantsequencesbyhybridization(dash):usingcas9toremoveunwantedhigh-abundancespeciesinsequencinglibrariesandmolecularcountingapplications.),《基因组生物学(genomebiol.)》2016;17:1–13。
2.wilsonmr,naccachesn,samayoae,biagtanm,bashirh,yug等人,通过新一代测序对神经钩端螺旋体病进行有指导意义的诊断(actionablediagnosisofneuroleptospirosisbynext-generationsequencing.),《新英格兰医学杂志(n.engl.j.med.)》2014;370:2408–17。
3.wilsonmr,shanbhagnm,reidmj,singhalns,gelfandjm,sampleha等人,利用宏基因组深度测序诊断巴氏阿米巴原虫性脑炎(diagnosingbalamuthiamandrillarisencephalitiswithmetagenomicdeepsequencing.),《神经学年刊(ann.neurol.)》2015;78:722–30。
4.rubyjg,bellarep,derisijl.,price:用于靶向组装(宏)基因组序列数据各组件的软件(price:softwareforthetargetedassemblyofcomponentsof(meta)genomicsequencedata.),g3(马里兰州贝塞斯塔(bethesdamd.),2013;3:865–80。
序列表
<110>crawford,emilyd
chow,ericd
derisi,josephl
<120>通过杂交找到低丰度序列(flash)的方法
<130>ucsf-546wo
<150>us62/375,789
<151>2016-08-16
<160>64
<170>patentin3.5版
<210>1
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>1
caaaacagcatagctctaaaacgatgttcgtgacggtttaaactatagtgagtcgtatta60
<210>2
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>2
caaaacagcatagctctaaaacttgaaccaaagttaccttggctatagtgagtcgtatta60
<210>3
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>3
caaaacagcatagctctaaaaccaagtcgcacgtacagtggtctatagtgagtcgtatta60
<210>4
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>4
caaaacagcatagctctaaaacaatgctgtgatcatttgacgctatagtgagtcgtatta60
<210>5
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>5
caaaacagcatagctctaaaaccttcaatcgcatcttcaagtctatagtgagtcgtatta60
<210>6
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>6
caaaacagcatagctctaaaaccgtccgttatccgttactttctatagtgagtcgtatta60
<210>7
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>7
caaaacagcatagctctaaaacagcaccataattatccgtgtctatagtgagtcgtatta60
<210>8
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>8
caaaacagcatagctctaaaactctgctttaaaacgaatgacctatagtgagtcgtatta60
<210>9
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>9
caaaacagcatagctctaaaactaactatttaagacacgcgtctatagtgagtcgtatta60
<210>10
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>10
caaaacagcatagctctaaaaccaagaatatgatacgccgccctatagtgagtcgtatta60
<210>11
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>11
caaaacagcatagctctaaaacttacaggcgtactgctgctgctatagtgagtcgtatta60
<210>12
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>12
caaaacagcatagctctaaaacgcgcagaagtactggccgcactatagtgagtcgtatta60
<210>13
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>13
caaaacagcatagctctaaaacatgctggcattggcgtacagctatagtgagtcgtatta60
<210>14
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>14
caaaacagcatagctctaaaacctgccgcagatttttatcctctatagtgagtcgtatta60
<210>15
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>15
caaaacagcatagctctaaaacgcgtgataaccatgcaacatctatagtgagtcgtatta60
<210>16
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>16
caaaacagcatagctctaaaacaccacatgaacgcggtaaatctatagtgagtcgtatta60
<210>17
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>17
caaaacagcatagctctaaaaccccatcagcatagtaataatctatagtgagtcgtatta60
<210>18
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>18
caaaacagcatagctctaaaactacaataacccgccgaaaccctatagtgagtcgtatta60
<210>19
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>19
caaaacagcatagctctaaaacactaaagcaagatttctaggctatagtgagtcgtatta60
<210>20
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>20
caaaacagcatagctctaaaacaagataaaaaagaacctctgctatagtgagtcgtatta60
<210>21
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>21
caaaacagcatagctctaaaacaattcgagtgctactctagcctatagtgagtcgtatta60
<210>22
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>22
caaaacagcatagctctaaaacaactttgtttttcgtgtcttctatagtgagtcgtatta60
<210>23
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>23
caaaacagcatagctctaaaactcatcatgtttggattatctctatagtgagtcgtatta60
<210>24
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>24
caaaacagcatagctctaaaacttaaatacaatttcttcattctatagtgagtcgtatta60
<210>25
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>25
caaaacagcatagctctaaaacaaactattatatatttttatctatagtgagtcgtatta60
<210>26
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>26
caaaacagcatagctctaaaaccttattttaatagttgtagtctatagtgagtcgtatta60
<210>27
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>27
caaaacagcatagctctaaaaccattgtttcggtctaaaattctatagtgagtcgtatta60
<210>28
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>28
caaaacagcatagctctaaaacctagaaaaagcgacttcacactatagtgagtcgtatta60
<210>29
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>29
caaaacagcatagctctaaaactcttttttctttttctctatctatagtgagtcgtatta60
<210>30
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>30
caaaacagcatagctctaaaacttgcgatgtactttcattgactatagtgagtcgtatta60
<210>31
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>31
caaaacagcatagctctaaaacgtagtatttatcggcatcatctatagtgagtcgtatta60
<210>32
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>32
caaaacagcatagctctaaaacgcgctgcttataccgctatactatagtgagtcgtatta60
<210>33
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>33
caaaacagcatagctctaaaactaacaagcacagctacaaaactatagtgagtcgtatta60
<210>34
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>34
caaaacagcatagctctaaaacaaccgaaaacaaaggcaagtctatagtgagtcgtatta60
<210>35
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>35
caaaacagcatagctctaaaacgctaatgaaacacctatagcctatagtgagtcgtatta60
<210>36
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>36
caaaacagcatagctctaaaacgaattgataatcgctgacatctatagtgagtcgtatta60
<210>37
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>37
caaaacagcatagctctaaaaccaaatataccgccacccgtactatagtgagtcgtatta60
<210>38
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>38
caaaacagcatagctctaaaacgtcttgcctgtttatttaaactatagtgagtcgtatta60
<210>39
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>39
caaaacagcatagctctaaaacaatgatgataaacgtgtcaactatagtgagtcgtatta60
<210>40
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>40
caaaacagcatagctctaaaacccgcgatacgtttcattcatctatagtgagtcgtatta60
<210>41
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>41
caaaacagcatagctctaaaactccacattggccattatgaactatagtgagtcgtatta60
<210>42
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>42
caaaacagcatagctctaaaacttagtaatgacaacgtttaactatagtgagtcgtatta60
<210>43
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>43
caaaacagcatagctctaaaacaataacattggtcgcttagactatagtgagtcgtatta60
<210>44
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>44
caaaacagcatagctctaaaactgctgctccaggccgctgagctatagtgagtcgtatta60
<210>45
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>45
caaaacagcatagctctaaaacgattcccctgcctgtgcggcctatagtgagtcgtatta60
<210>46
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>46
caaaacagcatagctctaaaacatctcgaacagggtctccggctatagtgagtcgtatta60
<210>47
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>47
caaaacagcatagctctaaaacctggcggtagtagtcgcggactatagtgagtcgtatta60
<210>48
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>48
caaaacagcatagctctaaaactagcgggcggtcgtccttgcctatagtgagtcgtatta60
<210>49
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>49
caaaacagcatagctctaaaacatggcgctgcaaccgcacagctatagtgagtcgtatta60
<210>50
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>50
caaaacagcatagctctaaaacgtacaacgtcgtattttatactatagtgagtcgtatta60
<210>51
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>51
caaaacagcatagctctaaaacccagctgcaatgcgttacacctatagtgagtcgtatta60
<210>52
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>52
caaaacagcatagctctaaaactgcttgttgtgctttaatcgctatagtgagtcgtatta60
<210>53
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>53
caaaacagcatagctctaaaacgccccgattgtgtggataatctatagtgagtcgtatta60
<210>54
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>54
caaaacagcatagctctaaaacaatcgcgccgctaatccgggctatagtgagtcgtatta60
<210>55
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>55
caaaacagcatagctctaaaacaggtcatacatagactcttcctatagtgagtcgtatta60
<210>56
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>56
caaaacagcatagctctaaaacacctactaaagcagttgctactatagtgagtcgtatta60
<210>57
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>57
caaaacagcatagctctaaaacgaaggctctaactctcttacctatagtgagtcgtatta60
<210>58
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>58
caaaacagcatagctctaaaacgcaataccaagtaaaaatcgctatagtgagtcgtatta60
<210>59
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>59
caaaacagcatagctctaaaaccgagtgagacttttcttttgctatagtgagtcgtatta60
<210>60
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>60
caaaacagcatagctctaaaacagcgcgctcacattcctgaactatagtgagtcgtatta60
<210>61
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>61
caaaacagcatagctctaaaacgtacctgagtacccatccaactatagtgagtcgtatta60
<210>62
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>62
caaaacagcatagctctaaaacgtgaatctgtttctgtcaatctatagtgagtcgtatta60
<210>63
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<400>63
caaaacagcatagctctaaaacatgacagcgatgccaagacgctatagtgagtcgtatta60
<210>64
<211>60
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸
<220>
<221>misc_feature
<222>(23)..(42)
<223>n是a、c、g或t
<400>64
caaaacagcatagctctaaaacnnnnnnnnnnnnnnnnnnnnctatagtgagtcgtatta60
- 还没有人留言评论。精彩留言会获得点赞!