一种用于纯化卡非佐米中间体的方法与流程
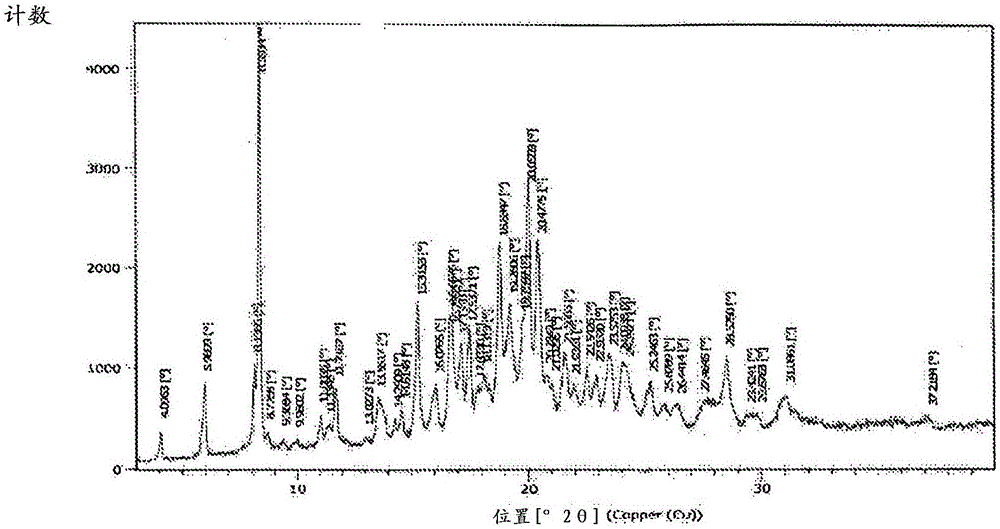
本发明涉及一种用于纯化卡非佐米(carfilzomib)合成中的中间体(即,式ii的化合物)的方法,其中x可以独立地选自三氟乙酸、盐酸、氢溴酸、对甲苯磺酸和磷酸,特别地降低了通常在式ii化合物合成过程中形成和/或存在的式iii杂质的水平,其中x可以独立地选自三氟乙酸、盐酸、氢溴酸、对甲苯磺酸和磷酸。本发明还涉及分离作为固体的式ii的化合物,优选式iia的化合物,及其用于制备卡非佐米的用途。本发明进一步涉及在8.39、15.31、17.13、18.83、20.05和20.47±0.2度2-θ具有x射线衍射峰的式iia的晶体化合物。其中x是三氟乙酸。
背景技术:
:卡非佐米,(2s)-n-{(1s)-1-苄基-2-[((1s)-3-甲基-1-{[(2r)-2-甲基环氧乙烷-2-基]羰基}丁基)氨基]-2-氧乙基}-4-甲基-2-({(2s)-2-[(吗啉-4-基乙酰基)氨基]-4-苯基丁酰基}氨基)戊酰胺,通过式i来表示:卡非佐米(cfz,以商品名kyprolis销售,onyxpharmaceuticals,inc.)是四肽环氧酮和选择性蛋白酶体抑制剂。它是环氧霉素(epoxomicin)的类似物。经美国食品药品管理局(fda)批准,与地塞米松联合、或与来那度胺加地塞米松联合,用于复发或难治性多发性骨髓瘤患者,所述患者已经接受一到三种治疗。它还被表明是治疗复发或难治性多发性骨髓瘤患者的单一药剂,所述患者已经接受一种或多种治疗。由式i表示的卡非佐米公开于美国专利no.7,417,042中。us7,417,042中所述的合成方案在以下描述为方案-1:方案-1其中,“boc”表示叔-丁氧基羰基;bz和bn分别表示苯甲酰基和苄基;mecn表示乙腈;tfa表示三氟乙酸;dmf表示二甲基甲酰胺;dcm表示二氯甲烷;diea表示二异丙基乙胺;hobt表示羟基苯并三唑;pybop表示苯并三唑-1-基-氧基三吡咯烷鏻六氟磷酸(oxytripyrrolidinophosphoniumhexafluorophosphate)。此外,美国专利7,231,818和8,207,297还描述了合成卡非佐米的相似方法。本发明的发明人发现上述方案形成了含有下文中所示的二醇卡非佐米杂质的卡非佐米:形成杂质的可能原因似乎是敏感性环氧环的打开。去除二醇卡非佐米杂质需要多重纯化,这随之降低了整体产量。多重纯化还可能导致敏感性环氧环的打开。wo2005/105827a2公开了将下文所示的式iva的化合物脱保护的方法:其中boc是叔-丁氧基羰基。式iva的化合物对应于wo2005/105827a2的化合物(cc)。在wo2005/105827a2中,在室温下用二氯甲烷中的三氟乙酸处理式iva的化合物一小时。将所得到的混合物浓缩并在真空下放置两小时。本发明的发明人发现上述方法导致下文中所示的式iiia杂质的形成:其中x是三氟乙酸。完成wo2005/105827a2中公开的方法后,通过高效液相色谱(hplc)测量了上述杂质为5.33面积%。进一步发现了上述方法形成了油形式的产物,其难以分离或纯化。一种用于合成式iia化合物的相似方法公开于wo2014/018807a1中。在wo2014/018807a1中,用二氯甲烷中的三氟乙酸在室温下处理式iva的化合物四小时。脱保护后,将反应混合物浓缩,以获得式iia的化合物。式iva和iia的化合物分别对应于wo2014/018807a1的化合物1068和1069。wo2014/018807a1的作者公开了按照以上方法获得的化合物是作为棕色油获得的。本发明的发明人发现按照wo2014/018807a1中公开的方法获得的化合物含有3.06%式iiia的杂质,如通过hplc测量的。通常,活性药物成分(api)中的杂质可能源自api自身的降解,或可以是过程产生的杂质。本发明的发明人发现在本发明的情况中,由于在卡非佐米制备的最后阶段中卡非佐米的环氧环打开形成了二醇卡非佐米杂质,或卡非佐米中间体中存在的开放环氧环的成分(或形式)可以与中间体一起形成二醇卡非佐米杂质。二醇卡非佐米杂质的去除非常困难,并且即使是通过使用不同的纯化方法,包括重结晶,最终化合物中仍然存在不理想含量的杂质。由于敏感性环氧环的存在,卡非佐米的多重纯化也是不理想的,所述环氧环在纯化过程中可能打开,形成二醇卡非佐米杂质。因此,需要在卡非佐米制备的中间阶段控制式iii的二醇杂质,以制备不含二醇卡非佐米杂质的卡非佐米。本申请的发明人已经发现了纯化式ii的化合物的方法,来制备基本上纯的式ii的化合物。特别地,式ii的化合物含有低于0.05面积%hplc的式iii的杂质。技术实现要素:在第一个方面中,本发明涉及用于纯化式ii的化合物的方法,其中x可以独立地选自三氟乙酸、盐酸、氢溴酸、对甲苯磺酸和磷酸。包括步骤:i)用合适溶剂中的碱金属高卤酸盐(perhalte)处理式ii的化合物,优选式iia的化合物;ii)添加抗溶剂;和ii)分离基本上纯的式ii或iia的化合物。本发明的另一个方面涉及式iia的晶体化合物。本发明的另一个方面涉及一种方法,包括将基本上纯的式ii或iia的化合物转化成卡非佐米。本发明的再一个方面涉及含有低于0.15面积%的二醇卡非佐米杂质的卡非佐米,如通过高效液相色谱(hplc)测量的。附图说明图1是式iia的晶体化合物的pxrd模式的说明。发明详述定义以下定义结合本申请来使用,除非上下文另有说明。如本文中使用的术语“胺保护基团”是指在分子的其它官能团或部分上进行反应时阻断(即,保护)胺功能性的基团。本领域技术人员将熟知胺保护基团的选择、连接和裂解并且将认识到许多不同的保护基团是本领域已知的,一个保护基团或另一个的适合性取决于计划的特定合成方案。有关该主题的论文可供参考,如greene和wuts,"protectivegroupsinorganicsynthesis,"第3版,pp.17-245(j.wiley&sons,1999),将其公开内容按引用并入本文中。术语碱金属专门是指锂(li)、钠(na)和钾(k)。碱金属高卤酸盐是指高碘酸锂、高碘酸钠、高碘酸钾、高溴酸锂、高溴酸钠,高溴酸钾、高氯酸锂、高氯酸钠和高氯酸钾。优选地,本发明提供了不含二醇杂质的卡非佐米。术语“不含二醇卡非佐米杂质的卡非佐米”是指含有低于0.15面积%的二醇卡非佐米杂质的卡非佐米,如通过高效液相色谱(hplc)测量的。更优选地,如本文中公开的卡非佐米含有低于0.10面积%的二醇卡非佐米杂质,如通过hplc测量的,并且最优选含有低于0.05面积%的二醇卡非佐米杂质,如通过hplc测量的。术语“基本上纯的式ii的化合物”是指含有低于0.15面积%的式iii杂质的式ii的化合物,如通过hplc测量的。更优选地,如本文中公开的,式ii的化合物含有低于0.10面积%的式iii的杂质,如通过hplc测量的,并且最优选含有低于0.05面积%的式iii的杂质,如通过hplc测量的。术语“式ii化合物或式iii化合物的x”涉及选自三氟乙酸、盐酸、氢溴酸、磷酸和对甲苯磺酸的相应盐。术语“x”类似于用于下文中表示的式iv的化合物的pg1保护基团的脱保护的脱保护酸的选择:其中pg1可以独立地选自叔-丁氧基羰基(boc)、芴甲氧基羰基(fmoc)、三苯甲基(trityl)、甲烷磺酰基(mesyl)和酰基。术语“约、一般、一般地”等解释为修饰术语或值,使其不是绝对的。这样的术语将由环境及其修饰的术语来定义,因为本领域技术人员理解这些术语。这至少包括用于测量值的给定技术的预期实验误差、技术误差和仪器误差的程度。如本文中使用的,术语“包括(comprising)”和“包括(comprises)”表示所述成分,或其结构或功能等同物,加上未列举的任何其它一个或多个成分。术语“具有”和“包括”也解释为开放式的。文中描述的所有范围包括端点,包括描述两个值之间的范围的那些。无论是否是所示的,本文所述的所有值都是由环境定义的近似值,包括用于测量值的给定技术的预期实验误差、技术误差和仪器误差的程度。术语“任选的”或“任选地”用于表示说明书中描述的事件或情况可能发生或可能不发生,并且描述包括其中事件发生的情况和事件未发生的情况。术语“抗溶剂”结合式ii的化合物的溶液时,是指降低式ii的化合物在溶液中的溶解度的液体,引起结晶或沉淀,在一些情况中自发地,而在其它情况中与另外的步骤,如放入晶种、冷却、划痕(scratching)和/或浓缩一起。在第一个方面中,本申请提供了用于纯化式ii化合物的方法,其中x可以独立地选自三氟乙酸、盐酸、氢溴酸、对甲苯磺酸和磷酸,通过用合适溶剂中的碱金属高卤酸盐处理,接着添加抗溶剂,来分离含有含量低于0.10面积%的式iii杂质的基本上纯的式ii的化合物,如通过高效液相色谱(hplc)测量的。碱金属高卤酸盐可以选自高碘酸锂、高碘酸钠、高碘酸钾、高溴酸锂、高溴酸钠,高溴酸钾、高氯酸锂、高氯酸钠和高氯酸钾。优选地,碱金属是高碘酸钠的水溶液。本发明的发明人发现使用碱金属高卤酸盐显著降低了式ii化合物中的式iii杂质的水平。特别地,式iii杂质降至低于0.05面积%hplc。发明人还发现了其它氧化剂,如高氯酸(hclo4)也可以有效地降低式iii杂质。在这个阶段,监测式iii杂质非常有利,因为它降低了二醇卡非佐米形成的含量。或者,还避免了为了除去二醇卡非佐米的卡非佐米的多重纯化。由于式iv化合物的脱保护,形成了式ii的化合物,相应的盐的形成取决于用于式iv化合物的pg1的脱保护的相应酸的使用。pg1是合适的胺保护基团。合适的胺功能保护基团以及保护和脱保护方法是本领域公知的(特别参见"protectivegroupsinorganicsynthesis",greenet.w.和wutsp.g.m.,wiley-interscience,1999)。优选地,合适的胺保护基团-pg1可以选自叔-丁氧基羰基(boc)、9-芴甲氧基羰基(fmoc)、三苯甲基(trityl)、甲烷磺酰基(mesyl)和酰基。使用胺脱保护的标准条件,通过裂解保护基团pg1,将胺保护的式iv的化合物转化成式ii化合物的相应盐,优选将合适的酸,如三氟乙酸、盐酸、氢溴酸、磷酸和对甲苯磺酸,用于合适的用于胺脱保护的溶剂中,如四氢呋喃、乙酸乙酯、二氯甲烷和乙腈。可以在降低或升高的温度下,例如,-30℃至40℃,更优选-10℃至10℃,在1分钟至10小时范围的时间段中,更优选30分钟至5小时,使用1至10当量的酸,更优选1至2当量,进行胺脱保护。在优选低于0℃的低温下进行脱保护反应是有利的。在低温下脱保护显著降低了式ii化合物的敏感性环氧环的打开,其转而在降低式iii杂质形成中是有用的。低温条件是有用的,但没有完全避免式iii杂质的形成。在优选实施方案中,任选在合适的溶剂(如二氯甲烷)的存在下,用三氟乙酸处理式iva的化合物,来制备式ii的化合物,其中x是三氟乙酸。将反应混合物在-5℃至0℃下搅拌1至10小时,更优选2至8小时。可以以固体分离式ii的化合物,或可以不进行分离就用于下一步使用碱金属高卤酸盐的纯化。优选地,通过使用本领域已知的合适技术来分离,如使用抗溶剂淬灭,接着沉淀,或通过从合适溶剂萃取,接着通过蒸发、蒸馏等从反应混合物除去溶剂,或可以使用任何其它方法。式ii化合物的分离是优选的,因为这降低了溶解的有机和/或无机杂质的含量。用合适溶剂中的碱金属高卤酸盐处理所获得的式ii的化合物(通常具有高于0.25面积%hplc的式iii的杂质),以除去式iii杂质。通过将反应混合物在5℃至40℃的温度范围下,更优选在20℃至30℃,搅拌2至15小时,更优选4至10小时,来进行纯化过程。纯化后,通过添加合适的抗溶剂来进行基本上纯的式ii的化合物的分离。可以通过将反应混合物加入抗溶剂中或通过将抗溶剂加入反应混合物中来实现抗溶剂的添加。添加可以是缓慢的或快速的。任选可以用合适的溶剂洗涤基本上纯的式ii的化合物,并且在合适的干燥条件下干燥。可以使用空气盘式干燥器、真空盘式干燥器、流化床干燥器、旋转闪蒸干燥器、闪蒸干燥器等中的任何一种来合适地进行干燥。可以在任何合适的温度下和大气压或高压下,或在减压下,进行干燥。在优选的实施方案中,基本上纯的式ii的化合物是晶体。用于式ii化合物纯化的合适溶剂可以选自四氢呋喃、二噁烷、二甲氧基乙烷、甲醇、乙醇、丙醇、异丙醇、正丁醇、乙酸乙酯、乙腈、丙腈、二甲基甲酰胺、二甲基乙酰胺和二甲基亚砜,或其混合物。优选地,所述溶剂是四氢呋喃。合适的抗溶剂可以选自水、烃,如正庚烷、正己烷、甲基叔-丁醚、二异丙醚。优选地,抗溶剂是水。优选的碱金属高卤酸盐可以选自以上讨论的碱金属高卤酸盐。最优选地,碱金属高卤酸盐是高碘酸钠。现有技术中没有报道式ii的化合物的分离,这使得难以去除杂质;然而,本发明的纯化方法涉及使用碱金属高卤酸盐,接着以晶体固体分离基本上纯的式ii的化合物,其中式iii的二醇杂质的含量降至低于0.05面积%hplc。优选地,本发明提供了含有低于0.15面积%二醇卡非佐米杂质的卡非佐米,如通过高效液相色谱(hplc)测量的。更优选地,卡非佐米含有低于0.10面积%的二醇卡非佐米杂质,如通过hplc测量的,并且最优选地,含有低于0.05面积%的二醇卡非佐米杂质,如通过hplc测量的。卡非佐米可以是晶体或无定形的。在一个实施方案中,将基本上纯的式ii的化合物,优选式iia的化合物,进一步用于卡非佐米的制备和分离,使用本领域已知的方法,如论文中公开的:‘journalofbiologicalchemistry’volume285no.51,40125-40134;2010。分离的卡非佐米优选不含二醇卡非佐米杂质,例如,含有低于0.15面积%,如通过hplc测量的。更优选地,分离的卡非佐米含有低于0.10面积%的二醇卡非佐米杂质,如通过hplc测量的,并且最优选含有低于0.05面积%的二醇卡非佐米杂质,如通过hplc测量的。分离的卡非佐米可以是晶体或无定形的。在以下实验部分中进一步给出了优选用于本
发明内容中的hplc方法。本发明的另一个方面提供了新的式iia的晶体化合物及其用于制备卡非佐米的用途。式iia的晶体化合物显示出在8.39、15.31、17.13、18.83、20.05和20.47±0.2度的折射2theta(θ)角的x-射线衍射峰;优选包括选自4.09、5.98、8.13、8.39、8.72、9.36、9.98、11.00、11.33、11.71、13.02、13.55、14.29、14.61、15.31、16.07、16.65、16.79、17.13、17.53、17.83、18.13、18.83、19.26、19.75、20.05、20.47、20.78、21.02、21.56、21.93、22.54、22.93、23.57、24.02、24.24、25.87、26.44、27.46、28.52、29.42、29.87、31.08和37.21±0.2度的折射2theta(θ)角的五个或更多个峰。x-射线粉末衍射(xrpd):在panalytical,model-empyreanx-ray粉末衍射仪上进行了xrpd分析。仪器参数如下所述:根据本发明的卡非佐米可以用于制备组合物,如药物组合物。在一个实施方案中,所述组合物包括通过根据本发明的方法生产的卡非佐米和药物学上可接受的赋形剂。药物学上可接受的赋形剂的非限制性实例包括稀释剂、盐、缓冲剂、ph调节剂、稳定剂、增溶剂、溶剂和防腐剂。在一个实施方案中,所述赋形剂是环糊精,如羟丙基-β-环糊精或磺丁基醚-β环糊精。实验通过以下实施例提供了根据本发明的详细实验参数,这些是为了说明而不是限制本发明的所有可能的实施方案。具体实施方案为了证明本申请的益处,进行了现有技术的实施例并且表示为参照实施例。实施例-1步骤a:式iia的晶体化合物的制备在三氟乙酸(720ml)中添加了式iva的化合物(170g),接着在-5℃下添加二氯甲烷(180ml)。将反应在相同温度下搅拌5小时。反应结束后,用水(5200ml)淬灭反应混合物。随后将反应过滤并用水(1800ml)洗涤,获得固体残余物(具有式iiia的杂质:0.43面积%hplc)。将所得到的化合物溶解于四氢呋喃(720ml)中,随后添加高碘酸钠溶液(180ml水中2.7gnaio4),并且在25-35℃下搅拌4小时。向所得到的搅拌过的溶液中,加入水(5220ml),获得标题化合物(178g)(具有式iiia的杂质:0.02面积%hplc)。实施例-2步骤a:式iia的晶体化合物的制备在三氟乙酸(640ml)中加入式iva的化合物(160g),接着在-5℃下添加二氯甲烷(640ml)。将反应在相同温度下搅拌5小时。反应完成后,用水(4800ml)淬灭反应混合物。随后将反应过滤并用水洗涤(两次,每次800ml),获得固体残余物(具有式iiia的杂质:0.29面积%hplc)。将所得到的化合物溶解于四氢呋喃(640ml)中,随后添加高碘酸钠溶液(160ml水中1.6gnaio4),并在25-35℃下搅拌4小时。向所得到的混合物中,加入水(4800ml),获得标题化合物(157g)(具有式iiia的杂质:0.04面积%hplc)。参照实施例参照实施例1(wo2005/105827a2的实施例10)式iia的化合物的制备在20-25℃下,将式iva的化合物(2g)加入三氟乙酸:二氯甲烷(8ml:2ml)混合物中。将反应搅拌1小时。搅拌后,将反应混合物在真空下浓缩,获得棕色油(具有式iiia的杂质:5.33面积%hplc)。参照实施例2(wo2014/018807的化合物1069的制备)式iia的化合物的制备在三氟乙酸(15ml)中添加式iva的化合物(5g),接着在20-25℃下添加二氯甲烷(15ml)。将反应在相同温度下搅拌4小时。反应完成后,将反应混合物浓缩,获得棕色油(具有式iiia的杂质:3.06面积%hplc)。通过高效液相色谱(hplc)测量了纯化式iia化合物后式iiia的杂质,并且将结果概括于下文的表-1中。表-1:使用了以下hplc方法:hplc系统配备有在210nm下运行的uv检测仪和hplc柱(ymcpackproc18rs(250×4.6)mm,5μm)。移动相为:移动相-a(mp-a):将2.72g磷酸二氢钾加入2l瓶中。添加2lhplc级水和2ml三乙胺。超声波处理,将物质完全溶解。使用磷酸,将溶液的ph调节至3.2±0.03。移动相-b(mp-b):乙腈。将移动相以(1.0ml/min)的流速泵过柱,柱温(25℃)和自动取样机温度(5℃)。mp-a和mp-b的混合物在(70分钟)运行过程中的梯度剖析如下:时间(min.)mp-a%mp-b%开始6535403565502575592575606535706535从表1的比较数据可以明显看出,在胺脱保护步骤中形成了高含量的式iiia的二醇杂质,其中在参考实施例1和2的现有技术方法中,二醇杂质的含量甚至更高。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1