在丝状真菌细胞中选择性碳源非依赖性表达蛋白质编码序列的方法与流程
本发明涉及一种在丝状真菌细胞中选择性碳源非依赖性表达蛋白质编码序列的方法,以及这种启动子用于在丝状真菌细胞中选择性碳源非依赖性表达蛋白质编码序列的用途。
背景技术:
丝状真菌广泛用于农业生物化学过程,比如纤维素或木质纤维素材料的水解,其例如在生物乙醇生产过程中用作生物质底物。此外,这种水解产物还可以作为生产许多有机化合物比如乳酸的底物,因此有效和可控的水解是现有技术中长期形成的需求。由于水解强烈依赖所述丝状真菌的水解酶的产生和所产生的酶的各自的产率或浓度,因此长期以来还需要在所需时间启动或停止某种酶的表达。
还需要使用强启动子,使得可以在某一时间获得足够水平的目标蛋白质。在本领域内,使用由例如li等(achievingefficientproteinexpressionintrichodermareeseibyusingstrongconstitutivepromoters;microbialcellfactories,2012,11:84)描述的组成型启动子。然而,优选使用所谓的诱导型启动子,其能够在某一时间启动或停止目标基因的转录,这通常与例如木质纤维素材料的复杂水解过程中的某些工艺步骤有关。由于源自丝状真菌的启动子通常不依赖于底物,因此难以控制转录,目前在实践中可用于在丝状真菌中控制目标基因转录的诱导性启动子只有相对有限的选择。如果使用的启动子不是底物非依赖性的,那么存在于所述底物中或在水解过程中由其他酶产生的化合物将影响酶的转录,使得控制变得不可能。
此外,这些启动子不仅需要是可控的,还需要表现出足够的表达效率和良好的表达率。理想地,表达应在诱导条件下达到高水平,并且应该能够在非诱导条件下被完全阻断。
技术实现要素:
现在本发明的发明人惊奇地发现这些需求可以通过在丝状真菌细胞中选择性碳源非依赖性表达蛋白质编码序列的方法来解决,所述方法包括以下步骤:
a)向丝状真菌细胞中引入长度为1550个碱基对(bp)的启动子,所述启动子包含序列模式x1x2x3x4tx5x6tcx7x8,其中x1为g、t或c,x2为a、g、t或c,x3为c或t,x4为c或t,x5为g、a、c或t,x6为c或t,x7为c或a,x8为c或t;
b)使含有所述启动子的所述丝状真菌细胞与生长培养基接触;
c)向所述生长培养基添加至少一种有机酸。
术语“碳源非依赖性表达”应理解为任何由化合物或物质启动的蛋白质编码序列的表达,所述化合物或物质不包含在用于所述丝状真菌生长的碳源(生长培养基)中或包含在用于所述丝状真菌生长的碳源(生长培养基)中但其浓度比所述碳源低至少两个数量级。
术语“表达”应理解为来自基因(在本申请中“所述蛋白质编码序列”)的信息用于合成功能性基因产物即蛋白质的过程。所述基因表达的过程被所有已知的生命——真核生物(包括多细胞生物)、原核生物(细菌和古细菌)采用,并被病毒利用——以生成生命所需的大分子机器。
在本发明的范围内,术语“碳源非依赖性表达”特别用于任何蛋白质的表达,所述蛋白质不是启动于或独立来自于用于丝状真菌细胞生长的各自底物的化合物,而且也不是启动于或独立来自于生长过程中产生的任何化合物,例如所述水解过程的中间产物。因此,如本发明方法的步骤b)中所定义,向生长培养基添加至少一种有机酸以启动蛋白质编码序列的转录。
在一种特别优选的实施方式中,所述有机酸选自抗坏血酸、叶酸、富马酸、苹果酸、丙二酸、泛酸、邻苯二甲酸、奎尼酸、琥珀酸、对苯二甲酸或其衍生物及其混合物。在本发明中,特别优选泛酸。
术语“蛋白质编码序列”应理解为涉及本领域技术人员已知的任何蛋白质编码序列。在一种特别优选的实施方式中,所述蛋白质为异源蛋白质。在一种进一步优选的实施方式中,所述蛋白质为来自于细菌或酵母微生物的异源蛋白质。在一种进一步特别优选的实施方式中,所述蛋白质编码序列为编码转录因子的酶的序列。在所述蛋白质编码序列是酶编码序列的情况下,所述序列优选选自酯酶、水解酶、裂解酶、氧化酶、重组酶和转移酶。
在本发明中,术语“丝状真菌细胞”应理解为来自天然存在和/或本领域技术人员已知的任何丝状真菌的任何细胞。该术语还包括天然来源或野生型或遗传修饰的任何丝状真菌细胞。在优选的实施方式中,所述丝状真菌细胞选自支顶孢菌属(acremonium)、曲霉属(aspergillus)、毛壳菌属(chaetomium)、镰刀菌属(fusarium)、腐质霉属(humicola)、耙菌属(irpex)、巨座殻属(magnaporte)、毁丝霉属(myceliophthora)、链孢霉属(neurospora)、青霉菌属(penicillium)、根霉属(rhizopus)、篮状菌属(talaromyces)、木霉属(trichoderma)和栓菌属(trametes),其中木霉属是特别优选的。
在根据本发明的方法中使用的启动子的长度为1550个碱基对(bp)并且包含序列模式x1x2x3x4tx5x6tcx7x8,其中x1为g、t或c,x2为a、g、t或c,x3为c或t,x4为c或t,x5为g、a、c或t,x6为c或t,x7为c或a,x8为c或t。在一种优选的实施方式中,以下位置优选选自:x1为c、x2为c、x3为t、x4为c、x5为a或c、x6为t或c、x7为a或c、x8为c。在一种特别优选的实施方式中,x5为c、x6为x和x7为c。
在另一种优选的实施方式中,所述启动子包含一个或多个如下碱基对:
在一种甚至进一步优选的实施方式中,所述启动子包含一个或多个以下碱基对:
在一种最优选的实施方式中,所述启动子选自由seqidno:1、seqidno:2、seqidno:3、seqidno:4和seqidno:5组成的组。
在将所述各自的启动子引入所述丝状真菌细胞后,根据本发明方法的步骤b)使所述丝状真菌细胞与生长培养基接触。“接触”可以通过适合于本发明目的的本领域技术人员已知的任何方法进行。在一种优选的实施方式中,向生长培养基添加所述丝状真菌细胞。所述生长培养基优选包含纤维素、木质纤维素和/或葡萄糖作为碳源。一个优选生长培养基的实施例是mandels-andreotti培养基。
根据本发明方法的步骤c),向所述生长培养基添加至少一种有机酸。根据步骤c)的“添加”可以通过本领域技术人员已知的适合于本发明目的的任何方法进行。所述至少一种有机酸优选以0.1至500mm,优选1至75mm,特别优选5至50mm的量添加。在选择里氏木霉作为丝状真菌细胞的情况下,在使用泛酸的情况下,所述至少一种有机酸以1至50mm,优选5至15mm的量添加,并且在使用奎尼酸的情况下,奎尼酸以10-500mm,优选50-150mm的量添加。
在进一步优选的实施方式中,所述启动子在载体内引入所述丝状真菌细胞中。在本发明中,术语“载体”用于定义任何能够将与报告基因融合的所述启动子序列稳定递送到所述丝状真菌细胞中的载体。优选的载体具有自身复制并转化到所述丝状真菌细胞中的能力,并且优选还包括可检测部分。优选的载体是,例如,质粒,其也可以在转化前进行去线性化(bedelinearized)或消化。
将所述启动子引入所述丝状真菌细胞根据本领域技术人员已知的适合于本发明目的的任何方法进行。这种方法例如在
在进一步的方面,本发明还涉及如前定义的启动子在丝状真菌细胞中选择性、碳源非依赖性表达蛋白质编码序列的用途。
附图说明
现在通过以下附图和实施例描述本发明。此处强调的是,附图和实施例不对本发明和权利要求书的范围构成限制,而仅仅构成对本发明、发明目的和通过本发明方法实现的益处的进一步说明。
附图说明:
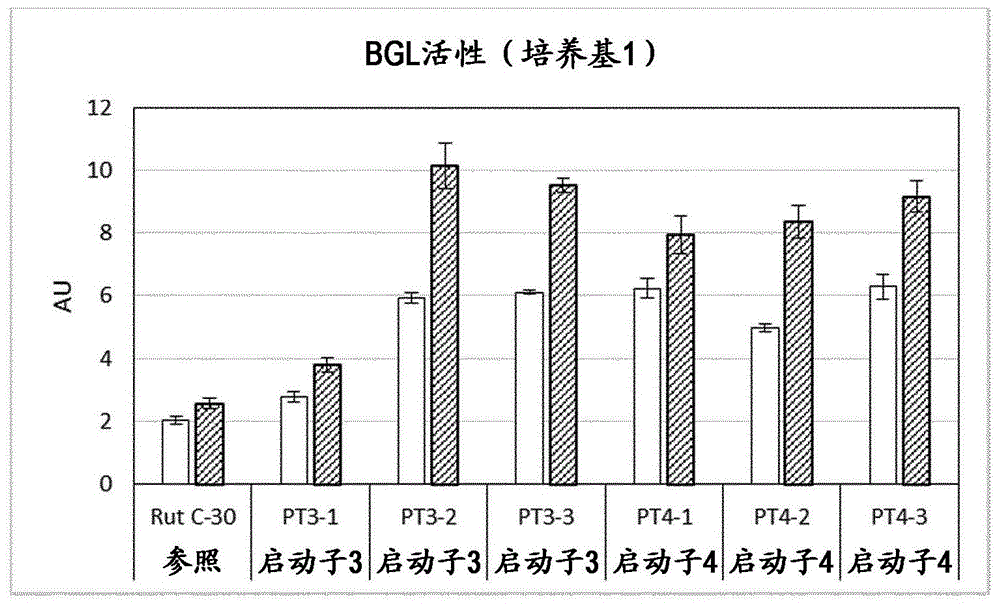
图1:显示培养基1中上清液的β-葡糖苷酶(bgl)测定的结果。白色条表示不添加泛酸作为诱导子的活性,阴影条表示将泛酸添加到各培养物的活性。y轴:如实施例1中所述计算的活性单位(au)。
图2:显示培养基2中上清液的β-葡糖苷酶(bgl)测定的结果。白色条表示不添加泛酸作为诱导剂的活性,阴影条表示将泛酸添加到各培养物的活性。y轴:如实施例1中所述计算的活性单位(au)。
具体实施方式
实施例1:
概述
所述实施例显示与不含所述启动子的各自的里氏木霉菌株的性能相比,均含有所述请求保护的序列模式的4种不同启动子在里氏木霉菌株中的性能,所述里氏木霉菌株在两种不同培养基中培养,以β-葡糖苷酶(seqidno:6)作为蛋白编码序列。
表达载体的构建
将本领域技术人员已知的且如例如sambrook和russel(molecularcloning–alaboratorymanual;coldspringharborlaboratorypress,newyork)或jansohn等(gentechnischemethoden,elsevier,münchen)所描述的标准方法用于dna琼脂糖凝胶电泳、dna纯化、大肠杆菌转化、质粒增殖和纯化、通过聚合酶链反应(pcr)扩增dna片段和从里氏木霉中分离基因组dna。基本上如aslanidis和dejong(1990,nucleicacidres.18(20),6069)所述进行连接非依赖性克隆(lic)。
按照制造商的说明,通过pcr扩增启动子1(seqidno:1),其使用来自里氏木霉的基因组dna作为模板,引物1fw(5’-actccatcactactgcttactattctcaaagggccgtttac-3’)和1rv(5’-ccaacttcctatacatgttcgaggcgtctccgctaatg-3’)和来自thermofisherscientific的phusion聚合酶(退火温度:72℃,延伸时间:45秒)。使用来自promega的wizardpcr纯化试剂盒纯化扩增子。
按照制造商的说明,通过pcr扩增启动子2(seqidno:2),其使用来自里氏木霉的基因组dna作为模板,引物2fw(5’-actccatcactactgattgcagacacttggacttg-3’)和2rv(5’-ccaacttcctatacatgcttgaaggagtgaagtagatagg-3’)和来自thermofisherscientific的phusion聚合酶(退火温度:60.9℃,延伸时间:25秒)。使用来自promega的wizardpcr纯化试剂盒纯化扩增子。
按照制造商的说明,通过pcr扩增启动子3(seqidno:3),其使用来自里氏木霉的基因组dna作为模板,引物3fw(5’-actccatcactactgcagaaccgtgccattttc-3’)和3rv(5’-ccaacttcctatacatggtgcatatcatgggatgag-3’)和来自thermofisherscientific的phusion聚合酶(退火温度:65.9℃,延伸时间:50秒)。使用来自promega的wizardpcr纯化试剂盒纯化扩增子。
按照制造商的说明,通过pcr扩增启动子4(seqidno:4),其使用来自里氏木霉的基因组dna作为模板,引物4fw(5’-actccatcactactgcttgaaggagtgaagtagatagg-3’)和4rv(5’-ccaacttcctatacatgattgcagacacttggacttg-3’)和来自thermofisherscientific的phusion聚合酶(退火温度:60.9℃,延伸时间:25秒)。使用来自promega的wizardpcr纯化试剂盒纯化扩增子。
按照制造商的说明,通过pcr扩增启动子5(seqidno:5),其使用来自里氏木霉的基因组dna作为模板,引物5fw(5’-actccatcactactgccaaatcgggcagag-3’)和5rv(5’-ccaacttcctatacatgcttgtgctgccaaggtgagag-3’)和来自thermofisherscientific的phusion聚合酶(退火温度:67.4℃,延伸时间:45秒)。使用来自promega的wizardpcr纯化试剂盒纯化扩增子。
按照制造商的说明用acui(来自newenglandbiolabs)消化并使用来自promega的wizardpcr纯化试剂盒纯化质粒ppromotortest(seqidno:7)。
使用连接非依赖性克隆(lic)将启动子1至5(如上所述通过pcr扩增)与线性化的ppromotortest融合。在dgtp存在下用t4dna聚合酶处理线性化载体。在dctp存在下用t4dna聚合酶处理扩增的启动子。如参考文献所述将t4dna聚合酶处理的载体和启动子混合并退火。然后将反应产物转化到化学感受态大肠杆菌xl1-blue细胞中,铺板在含有100mg·1-1氨苄青霉素(lb-amp)的lb-琼脂平板上,并在37℃下孵育24小时。使用牙签从琼脂平板挑取菌落,转移到液体lb-amp培养基中并在37℃下振荡(250rpm)孵育24小时。按照制造商的说明分离质粒dna并通过pcr验证插入物的整合,其使用引物testfw(5’-gagcaatgtgggactttgatg-3’)和testrv(5’-gcccaatcttgggatgctac-3’)和来自promega的gotaqgreenmastermix(退火温度:60℃,延伸时间:2分45秒)。通过测序验证得到预期条带(启动子1:2.5kb,启动子2:1.8kb,启动子3:2.7kb,启动子4:1.8kb,启动子5:2.6kb,空载体:1.1kb)的质粒并分别命名为pptest-1至-5。
将所述启动子测试载体转化到里氏木霉
按照制造商的说明,用sbfi(来自newenglandbiolabs)消化载体pptest-1至-5。基本上如
从菌株的菌丝体中分离基因组dna。按照制造商的说明,通过pcr进行扩增重组启动子区,其使用基因组dna为模板,引物intfw(5’-gcacaaccgcatgatataggg-3’)和intrv(5’-cccaatcttgggatgctacc-3’)和来自thermofisherscientific的phusion聚合酶(退火温度:65.7℃,延伸时间:1分钟35秒)。使用来自promega的wizardpcr纯化试剂盒纯化扩增子并测序。
将含有载体pptest-1的菌株命名为pt1-1至-3。
将含有载体pptest-2的菌株命名为pt2-1至-3。
将含有载体pptest-3的菌株命名为pt3-1至-3。
将含有载体pptest-4的菌株命名为pt4-1至-3。
将含有载体pptest-5的菌株命名为pt5-1至-3。
在摇瓶中生长报告菌株
菌株于摇瓶中在以下两种培养基中生长:
培养基1含有(g·l-1):(nh4)2so41.4、kh2po42.0、mgso4*7h2o0.3、cacl2*2h2o0.3、邻苯二甲酸钾10.2、feso4*7h2o0.005、mnso4*h2o0.0016、znso4*7h2o0.0014、cuso4*5h2o0.0001、avicel(=微晶纤维素)20和来自提取的大豆的研磨残余物5。用hcl或naoh将培养基调节至ph5。
培养基2含有(g·l-1):(nh4)2so42.8、kh2po42.0、mgso4*7h2o0.3、cacl2*2h2o0.3、邻苯二甲酸钾10.2、feso4*7h2o0.02、mnso4*h2o0.0064、znso4*7h2o0.0056、cuso4*5h2o0.0004、葡萄糖10和酵母提取物0.5。用hcl或naoh将培养基调节至ph5.5。
在无菌罩下将15ml培养基分配到50ml锥形摇瓶中,并用橡胶泡沫盖封闭该烧瓶。解冻所述转化子和菌株rutc-30的分生孢子原种,并在无菌罩下将75μl分生孢子悬浮液移液到含所述培养基的锥形瓶中。每种菌株和培养基接种六个烧瓶。将烧瓶在30℃下振荡(250rpm)孵育6天(培养基1)或3天(培养基2)。为了测试启动子的诱导,在48(培养基1)或24小时(培养基2)后,将10mm泛酸加入到每种菌株和培养基的三个烧瓶中。
将培养物倒入15ml管中,离心(3220xg,4℃,15分钟),将上清液转移到新管中并在4℃下储存。
分析培养上清液中的β-葡糖苷酶活性
将β-葡糖苷酶(bgl)用作报告基因以测试启动子活性,因此测量培养上清液的bgl活性以测定整合在各菌株中的启动子的活性。为了测量活性,如果需要,首先将上清液在乙酸盐缓冲液(50mm;ph5.0)中稀释。将50μl样品与50μl2mm对硝基苯酚-β-d-吡喃葡萄糖苷(溶于乙酸盐缓冲液中)混合,并在60℃下孵育30分钟。然后加入100μl1mna2co3(溶于水中),并使用分光光度计测量405nm处的吸收。通过乘以吸光度值(a405)和稀释度来计算活性单位(au)。
结果显示在图1和2中。
从图和实施例中可以看出,包含本发明序列模式的所有测试启动子都满足请求保护的益处并且在诱导条件下导致显著改进的目标蛋白质的表达。
- 还没有人留言评论。精彩留言会获得点赞!