序列变体的检测的制作方法
本公开内容一般地涉及用于通过核酸扩增来检测序列变体的系统和方法。
背景技术:
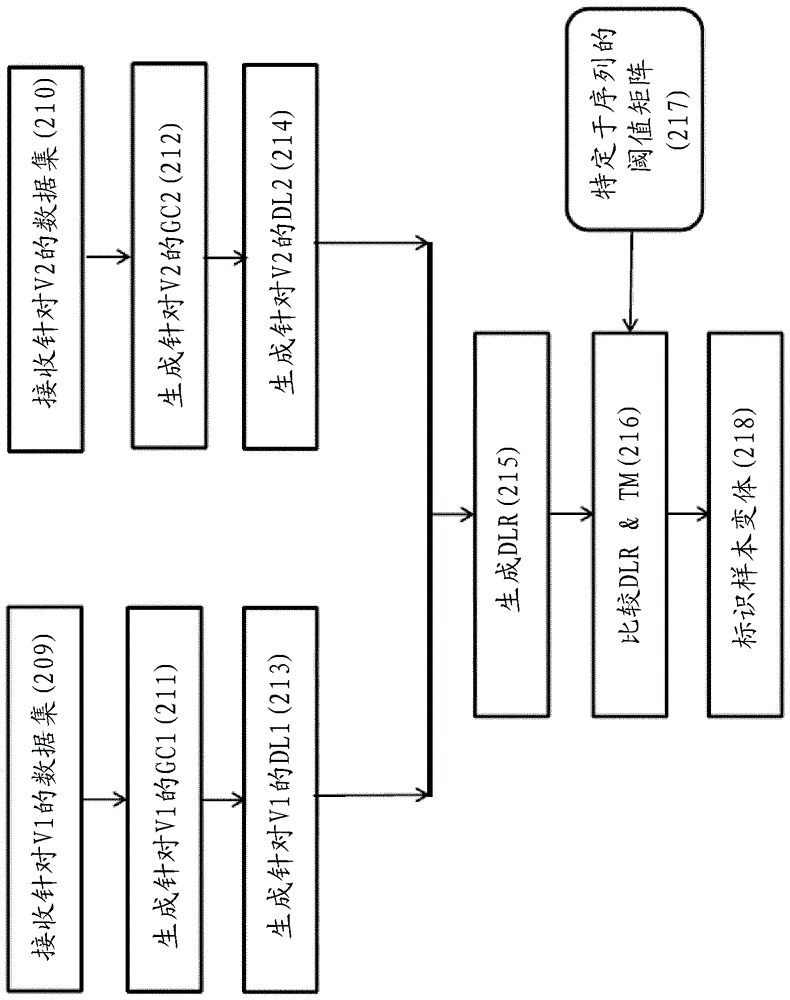
:所有生物体(例如动物、植物和微生物)的遗传信息被编码在脱氧核糖核酸(dna)中。在人类中,完整的基因组包含位于24条染色体上的大约100,000个基因(人类基因组,t.strachan,bios科学出版社,1992)。每个基因为特定的蛋白质编码,所述每个基因在其经由转录和翻译的表达之后履行活细胞内的特定生物化学功能。遗传密码中的改变或变化可导致mrna的表达序列或水平中的以及潜在地在通过mrna所编码的蛋白质中的改变。这些改变,已知为多态性或突变,可对导致疾病的mrna或蛋白质的生物活性具有显著不利影响。突变包括核苷酸删除、插入、替换或其它变更(即点突变)。由遗传多态性所引起的许多疾病是已知的并且包括血友病、地中海贫血、杜氏肌营养不良(dmd)、亨廷顿氏舞蹈病(hd)、阿耳滋海默氏病以及囊胞性纤维症(cf)(humangenomemutations,d.n.cooper和m.krawczak,bios出版社,1993)。诸如这些之类的遗传疾病可由形成特定基因的dna中的单个核苷酸的单个添加、替换或删除造成。除了导致遗传疾病的突变基因之外,某些先天缺陷是染色体异常的结果,所述染色体异常诸如21三体(唐氏综合征)、13三体(帕陶综合征)、18三体(爱德华氏综合征)、x单体性(特纳氏综合征)以及其它性染色体异倍性、诸如克氏综合征(xxy)。此外,存在如下的增长的证明:某些dna序列可使个体偏向于多个疾病中的任何,所述疾病诸如糖尿病、动脉硬化、肥胖、各种自体免疫疾病和癌症(例如结肠直肠的、乳房的、卵巢的、肺的)。某些多态性被认为使某些个体偏向于疾病或有关于某些疾病的发病率水平。动脉粥样硬化、肥胖、糖尿病、自体免疫失调和癌症是被认为具有与多态性的相关性的一些这样的疾病。除了与疾病的相关性之外,多态性还被认为在患者对被给予用于治疗疾病的治疗剂的响应中起作用。例如,相信多态性在患者响应于药物、放射治疗和其它形式的治疗的能力中起作用。标识多态性可导致对特定疾病的更好理解以及针对这样的疾病的潜在更有效的治疗。的确,基于患者的经标识的多态性的经个性化的治疗方案(therapyregiment)可导致拯救生命的医学干预。一旦多态性被标识并且被隔离,就可以发现新药物或化合物,其与特定多态性的产物相互作用。基于多态性还可以实现对包括病毒、细菌、朊病毒和真菌之类的传染性有机体(organism)的标识,并且适当的治疗响应可以被施予受感染的主体。由于大约16个核苷酸的序列在统计地面上是特定的,甚至是对于人类基因组的大小而言,因此相对短的核酸序列可以用于检测高等生物中的正常和缺陷基因,并且用于检测传染性的微生物(例如细菌、真菌、原生生物和酵母)以及病毒。dna序列还可以用作用于检测相同物种内的不同个体的指纹(参见thompson、j.s.和m.w.thompson、eds.,geneticsinmedicine,w.b.saundersco.,费城,pa.(1991))。已经开发了众多方法来检测和分析核酸序列。例如,可以通过将经扩增的核酸分子的移动性与通过凝胶电泳的已知标准相比较,或者通过利用与待标识序列互补的探针的杂交,来标识核酸序列。用于分析核酸序列的领域公认的方法采用聚合酶链反应(polymerasechainreaction,pcr),用于酶促地合成或扩增所定义的核酸序列的体外方法。所述反应通常使用两个寡核苷酸引物,所述寡核苷酸引物杂交到相对的链并且位于待扩增的模板或目标dna序列的侧面。通过热稳定的dna聚合酶来催化引物的伸长。涉及模板变性、引物退火以及通过聚合酶对经退火引物的扩展的循环的重复序列提供特定dna片段的指数式累积。在用于促进扩增过程的检测和量化的过程中通常使用荧光探针或标记。在典型的动力学pcr分析中,相对于典型pcr过程的循环数目来绘制荧光强度值,并且在pcr过程的每个循环中监视pcr产物的形成。通常在热循环器中测量扩增,所述热循环器包括用于在扩增反应期间测量荧光信号的部件和设备。这样的热循环器的示例是罗氏诊断光循环器(cat.no.20110468)。例如如下来检测扩增产物:借助于经荧光标记的杂交探针,所述经荧光标记的杂交探针仅仅在它们被结合到目标核酸的时候发射荧光信号,或在某些情况中还借助于结合到双链dna的荧光染料。然而,在由扩增反应所产生的数据中可出现误差,从而引起数据分析以及从分析中获得的作为结果的物理性质(例如起始材料的标识和/或量化)中的误差。此外,当分析大群核酸中的相对小的序列变化的时候,这些误差可混合。因此,标识用于通过使用核酸扩增方法的输出来分析序列变体信息的经改进的过程将是有益的。技术实现要素:在一个方面中,提供用于检测样本中至少两个核酸序列变体的方法,所述方法包括步骤:(a)扩增所述至少两个序列变体以产生包括第一变体扩增子和第二变体扩增子的至少两个扩增产物,其中所述扩增步骤相应地为第一和第二扩增子产出第一和第二增长曲线;(b)通过使用处理器通过如下来分析第一和第二增长曲线从线性的相对偏离:生成第一增长曲线从线性的第一偏离以及第二增长曲线从线性的第二偏离,并且比较所述从线性的第一和第二偏离以生成从线性比的偏离;(c)通过使用处理器来比较从线性的相对偏离与阈值矩阵;以及(d)通过使用处理器基于所述比较步骤(c)的结果来标识所述两个或更多序列变体。在一些实施例中,所述分析步骤(b)包括计算针对第一增长曲线的deltab1并且计算针对第二增长曲线的deltab2,其中deltabn=(最大|ŷi-yi|/广义基线平均数);并且从线性的相对偏离=deltab1/deltab2。在一些实施例中,从线性的相对偏离包括log10(deltab1/deltab2)。在一些实施例中,deltabn=(最大|ŷi-yi|/基线截距)。在本文中,在一些实施例中,基线截距包括y中位数(ymedian)、几何平均数、调和平均数或广义平均数。在某些实施例中,基线截距包括y中位数。在一些实施例中,deltabn=(最大|ŷi-yi|/y中位数)。在一些实施例中,根据循环m到p来评估从线性的偏离,其中0<m<p。在某些实施例中,m>2。在某些实施例中,3<m<7。在某些实施例中,m=6。在另一方面中,提供一种用于检测样本中的两个或更多核酸序列变体的系统,所述系统包括可操作地被连接到存储器、处理器和显示器的核酸扩增模块,其中所述处理器被配置成执行用于检测两个或更多序列变体的计算机实现的方法,所述方法包括:(a)通过使用核酸扩增模块来扩增所述至少两个序列变体,用于产生包括第一变体扩增子和第二变体扩增子的至少两个扩增产物,(b)通过使用处理器来相应地为第一和第二扩增子生成第一和第二增长曲线;(c)通过使用处理器通过如下来分析第一和第二增长曲线从线性的相对偏离:生成第一增长曲线从线性的第一偏离以及第二增长曲线从线性的第二偏离,并且比较所述从线性的第一和第二偏离以生成从线性比的偏离;(d)通过使用处理器来比较从线性的相对偏离与阈值矩阵;以及(e)通过使用处理器基于所述比较步骤(d)的结果来标识所述两个或更多序列变体。在一些实施例中,所述分析步骤(b)包括计算针对第一增长曲线的deltab1并且计算针对第二增长曲线的deltab2,其中deltabn=(最大|ŷi-yi|/广义基线平均数);并且从线性的相对偏离=deltab1/deltab2。在一些实施例中,从线性的相对偏离包括log10(deltab1/deltab2)。在一些实施例中,deltabn=(最大|ŷi-yi|/基线截距)。在本文中,在一些实施例中,基线截距包括y中位数、几何平均数、调和平均数或广义平均数。在某些实施例中,基线截距包括y中位数。在一些实施例中,deltabn=(最大|ŷi-yi|/y中位数)。在一些实施例中,根据循环m到p来评估从线性的偏离,其中0<m<p。在某些实施例中,m>2。在某些实施例中,3<m<7。在某些实施例中,m=6。此外,本公开内容提供了一种用于检测样本中的两个或更多核酸序列变体的系统,所述系统包括可操作地被连接到存储器、处理器和显示器的核酸扩增模块。所述系统被配置成通过如下来执行用于检测所述两个或更多序列变体的方法:通过使用核酸扩增模块来扩增所述至少两个序列变体以产生包括第一变体扩增子和第二变体扩增子的至少两个扩增产物,并且通过使用处理器来相应地为第一和第二扩增子生成第一和第二增长曲线。通过使用处理器来分析增长曲线以通过如下来确定第一和第二增长曲线从线性的相对偏离:生成第一增长曲线从线性的第一偏离以及第二增长曲线从线性的第二偏离,并且比较所述第一和第二偏离以生成从线性比的偏离。通过使用处理器,所述系统然后比较从线性的相对偏离与阈值矩阵,并且从而标识所述两个或更多序列变体。而且,提供了一种用于检测样本中的两个或更多核酸序列变体的系统,其中所述系统包括可操作地被连接到存储器、处理器和显示器的核酸扩增模块。所述核酸扩增模块扩增所述至少两个序列变体以产生包括第一变体扩增子和第二变体扩增子的至少两个扩增产物,并且通过使用处理器相应地为第一和第二扩增子生成第一和第二增长曲线。此后,所述处理器被配置成接收第一数据集,所述第一数据集表示针对第一核酸变体扩增的增长曲线;接收第二数据集,所述第二数据集表示针对附加核酸变体扩增的增长曲线,其中所述附加核酸变体包括在相对于第一核酸变体的多态位点处的所述至少一个序列修改;生成拟合所述第一数据集的第一曲线以及拟合所述第二数据集的第二曲线;生成第一曲线从线性的第一偏离以及第二曲线从线性的第二偏离;比较从线性的第一偏离与从线性的第二偏离以标识从线性比的偏离;比较从线性比的偏离与阈值矩阵;以及基于与阈值矩阵的比较来标识所述两个或更多序列变体。此外提供了一种用于检测样本中至少两个核酸序列变体的方法,所述方法包括步骤:扩增所述至少两个序列变体,用于产生包括第一变体扩增子和第二变体扩增子的至少两个扩增产物,其中所述扩增步骤相应地为第一和第二扩增子产出第一和第二增长曲线;通过使用处理器通过如下来分析第一和第二增长曲线从线性的相对偏离:生成第一增长曲线从线性的第一偏离以及第二增长曲线从线性的第二偏离,并且比较所述从线性的第一和第二偏离以生成从线性比的偏离;通过使用处理器来比较从线性的相对偏离与阈值矩阵;以及通过使用处理器基于比较步骤的结果来标识所述两个或更多序列变体。还提供的是一种用于检测两个或更多序列变体的计算机实现的方法,所述方法包括在计算机系统处:接收第一数据集,所述第一数据集表示针对第一核酸变体扩增的增长曲线;接收第二数据集,所述第二数据集表示针对附加核酸变体扩增的增长曲线,其中所述附加核酸变体包括在相对于第一核酸变体的多态位点处的所述至少一个序列修改;生成拟合所述第一数据集的第一曲线以及拟合所述第二数据集的第二曲线;生成第一曲线从线性的第一偏离以及第二曲线从线性的第二偏离;比较从线性的第一偏离与从线性的第二偏离以标识从线性比的偏离;比较从线性比的偏离与阈值矩阵;以及基于所述比较步骤来标识所述两个或更多序列变体。参考以下具体实施方式和附图可以获得对本公开内容的性质和优点的更好的理解。附图说明图1是如本文中所述的系统的框图。图2(a)是所述系统所采用的方法的示意性图示。图2(b)是所述方法的数据处理部分的示意性图示。图3是序列特定的阈值矩阵的使用的特定示例的图示。图4图示了常规序列变体逻辑的特定示例。图5是针对核酸扩增增长曲线的deltab的图形描绘。图6图示了经改进的序列变体方法。图7示出了通过使用新方法所获得的结果。图8示出了通过使用常规序列变体逻辑的对假gg等位基因的标识。图9示出了通过使用常规序列变体逻辑,由于cy5.5或famrfi(仅仅aa)降落到截止以下,某些样本是无效的。图10示出了通过使用常规序列变体逻辑,由于deltact值在范围外部,某些ga试样是无效的。图11示出了aa试样如何由于hexrfi值在截止以上而是无效的。然后将样本视为ga样本,因为所有通道(fam、hex和cy5.5)ct存在,但是未通过δct截止。具体实施方式实施例可以基于核酸扩增反应来检测序列变体。常规的方法不准确地标识序列变体,然而本文中所述的经改进的方法提供一种机制来一致地并且准确地标识没有通过使用常规方法被正确检测的变体。此外,已经发现当评估有挑战性的样本的时候、例如在存在潜在抑制性组分的情况中,经改进的方法是更稳健的。除非在本文中另行定义,否则本文中所使用的科学和技术术语具有本领域中的普通技术人员所通常理解的含义。此外,除非由上下文另行要求,否则单数术语包括复数并且复数术语包括单数。冠词“一”和“一个”在本文中被使用以指代一个或多于一个(即指代至少一个)该冠词的语法对象。作为示例,“元件”意指一个元件或多于一个元件。术语“检测”、“在检测”、“检测到”以及类似的术语在本申请中被使用来宽泛地指代过程或者发现或确定存在或不存在、以及程度、数量或水平、或某物出现的概率。例如,当参考目标核酸序列被使用的时候的术语“在检测”可以表示对存在、不存在的发现或确定、水平或数量、以及序列存在或不存在的概率或似然性。将理解到,表述“在检测存在或不存在”、“检测到存在或不存在”以及有关的表述包括定性和定量检测。术语“核酸”、“多核苷酸”以及“寡核苷酸”是指核苷酸的聚合物(例如核糖核苷酸或脱氧核苷酸),并且包括自然出现的(腺苷、胍、胞嘧啶、尿嘧啶和胸苷)、非自然出现的以及经修改的核酸。术语不受聚合物的长度(例如单体数量)限制。核酸可以是单链或双链的,并且将一般包含5’-3’磷酸二酯键,尽管在一些情况中,核苷酸类似物可具有其它键合(linkage)。单体典型地被称为核苷酸。术语“非自然核苷酸”或“经修改的核苷酸”是指如下核苷酸:所述核苷酸包含经修改的含氮碱基、糖或磷酸基团,或在其结构中并入了非自然的半部(moiety)。非自然核苷酸的示例包括双脱氧核苷酸、生物素化的、胺化了的、脱氨基的、烷基化的、苯甲基化的以及经荧光体标记的核苷酸。可以包括对本文中所述的标准的使用的合适核酸扩增方法包括但不限于连接酶链反应(lcr;wud.y.和wallacer.b.,genomics4(1989)560-69;以及baranyf.,proc.natl.acad.sci.usa88(1991)189-193);聚合酶连接酶链反应(baranyf.,pcrmethodsandapplic.1(1991)5-16);gap-lcr(wo90/01069);修复链反应(ep0439182a2),3sr(kwohd.y.等人,proc.natl.acad.sci.usa86(1989)1173-1177;guatellij.c.等人,proc.natl.acad.sci.usa87(1990)1874-1878;wo92/08808),以及nasba(us5,130,238)。此外,还可以使用以下方法中的一个或多个:链置换扩增(stranddisplacementamplification,sda)、转录介导扩增(transcriptionmediatedamplification,tma)以及qb-扩增(为了回顾,参见例如whelena.c.和persingd.h.,annu.rev.microbiol.50(1996)349-373;abramsonr.d.和myerst.w.,curropinbiotechnol4(1993)41-47)。如上所述,在特定的实施例中,通过pcr来分析核酸序列,所述pcr除了其它参考文献之外尤其在例如专利号为4,683,202、4,683,195、4,800,159和4,965,188的美国专利中被公开。pcr典型地采用两个或更多寡核苷酸引物,所述寡核苷酸引物结合到所选核酸模板(例如dna或rna)。对于核酸分析有用的引物包括能够充当目标核酸的核酸序列内的核酸合成的发起点的寡核苷酸。可以通过常规方法来从限制酶切消化中纯化引物,或可以合成地产生它。为了扩增的最大效率,引物优选是单链的,但是引物可以是双链的。双链引物首先被变性,即被处理以使链分离。用于使双链核酸变性的一种方法是通过加热。“热稳定的聚合酶”是一种热稳定的聚合酶,即它是如下一种酶:其催化与模板互补的引物扩展产物的形成,并且当在对于实现双链模板核酸的变性所必要的时间内经受升高的温度的时候不是不可逆地变性。一般,合成在每个引物的3’端处被发起,并且在沿着模板链的5’到3’方向上继续进行。热稳定的聚合酶已经例如与以下各项隔离:黄栖热菌属、红栖热菌属、嗜热栖热菌属、水生栖热菌属、乳色栖热菌属、鲁宾斯栖热菌属、嗜热脂肪芽孢杆菌、以及甲烷嗜热菌。尽管如此,假如酶被补给,则在pcr试验中还可以采用不是热稳定的聚合酶。如果模板核酸是双链的,则在它可以被用作pcr中的模板之前有必要使两个链分离。可以通过任何合适的变性方法来实现链分离,包括物理、化学或酶促手段。用于分离核酸链的一种方法涉及加热核酸直到它占绝大多数地被变性为止。对于使模板核酸变性而言必要的加热条件将取决于例如缓冲器盐浓度以及在被变性的核酸的长度以及核苷酸组成,但是在取决于反应特征、诸如温度以及核酸长度的时间内通常范围从大约90℃到大约105℃。变性通常被执行大约5秒到9分钟。如果通过热来使双链模板核酸变性,则允许反应混合物冷却到如下温度:所述温度促进每个引物退火至其在目标核酸上的目标序列。用于退火的温度优选地从大约35℃到大约70℃,此外优选地大约45℃到大约65℃;此外优选地大约50℃到大约60℃,此外优选地大约55℃到大约58℃。退火时间可以从大约10秒到大约1分钟(例如大约20秒到大约50秒;大约30秒到大约40秒)。反应混合物然后被调节到如下温度:在所述温度下,聚合酶的活性被促进或优化,即对于从经退火的引物发生扩展以生成与待分析的核酸互补的产物而言足够的温度。温度应当足以从每个引物合成扩展产物,所述每个引物被退火至核酸模板,但是不应当如此高使得来自其互补模板的扩展产物变性(例如用于扩展的温度一般范围从大约40°到80℃(例如大约50℃到大约70℃;大约60℃)。扩展时间可以从大约10秒到大约5分钟,优选地大约15秒到2分钟,此外优选地大约20秒到大约1分钟,此外优选地大约25秒到大约35秒。新合成的链形成双链分子,所述双链分子可以被使用在反应的随后的步骤中。链分离、退火和伸长的步骤可以如对于产生所期望的量的与目标核酸相对应的扩增产物而言所需要的那样经常地被重复。反应中的限制因素是反应中存在的引物、热稳定的酶、以及核苷三磷酸的量。循环步骤(即变性、退火和扩展)优选地被重复至少一次。为了在检测中使用,循环步骤的数目将取决于例如样本的性质。如果样本是核酸的复杂混合物,则将需要更多循环步骤来扩增足够用于检测的目标序列。一般,循环步骤被重复大约20次,但是可以被重复少于20次或者如40、60或甚至100次那么多。此外,可以实施pcr试验,其中在相同的步骤(一步pcr)中或在分离的步骤(两步pcr)中执行退火和扩展的步骤。术语“样本”是指来自个体的包含或假定包含核酸的任何组成。术语包括细胞、组织、或血液和血液组分的经纯化或分离的组分,例如dna、rna、蛋白质、无细胞的部分、细胞溶解产物、血浆、血小板、血清、白膜层以及干血斑点。样本还可以包括从个体获得的细胞的体外培养的成分和组分,包括细胞系。样本的特定附加示例包括排泄物、粘膜棉签、组织抽出物、组织匀浆、细胞培养物、以及细胞培养物浮层(包括真核以及原核细胞的培养物)、尿液、唾液、痰以及脑脊髓样本。本文中所述的方法可以用于标识核酸序列变体。可以标识各种类型的序列变体,包括但不限于等位基因、多态性、单倍型和/或基因型。术语“等位基因”是指基因的序列变体。一个或多个遗传差异可以构成等位基因。术语“多态性”是指如下状况:其中在种群中可以发现特定核苷酸序列的两个或更多变体。多态定位是指核酸序列中的一位点,在该处出现区分变体的多态核苷酸。“单个核苷酸多态性”或snp是指由单个核苷酸组成的多态位点。术语“单倍型”是指个体中相同染色体上的不同位置(基因座或基因)处的等位基因的组合,而术语“基因型”是指在个体或样本中所包含的基因的等位基因的总和。在本文中所述的方法中,可以通过检测多态位点中的一个或多个处存在的核苷酸来标识序列变体。可以使用包含感兴趣的(一个或多个)核酸的任何类型的组织。在本领域中已知多种方法来用于标识序列异常,并且所使用的特定方法不是公开内容的关键方面。示例性的方法涉及特定于等位基因的扩增,或对经扩增的核酸的基于探针的检测。在一个实施例中,可以通过使用特定于等位基因的扩增或引物扩展方法来标识等位基因,所述方法基于末端引物失配对于dna聚合酶扩展引物的能力的抑制作用。为了通过使用特定于等位基因的扩增或基于扩展的方法来检测等位基因序列,选择与每个等位基因互补的引物使得3'末端核苷酸在多态定位处杂交。在存在目标等位基因的情况中,引物匹配3'末端处的目标序列,并且引物被扩展。在存在另一等位基因的情况中,引物具有相对于目标序列的3'失配,并且引物扩展要么被消除要么被显著缩减。在例如专利号为5,137,806;5,595,890;5,639,611的美国专利以及专利号为4,851,331的美国专利中描述了特定于等位基因的扩增或基于扩展的方法。用于基因型分型的基于特定于等位基因的扩增的方法可以促进对单倍型的标识。本质上,特定于等位基因的扩增用于扩增来自杂合样本中两个等位基因之一的、包括多个多态位点的区域。经扩增的序列内存在的snp变体然后被标识,诸如通过探针杂交或定序。在另一实施例中,使用基于探针的方法,其依赖于在探针及其包括多个目标等位基因的对应目标序列之间所形成的杂交双重物(duplexe)的稳定性中的差异。在充分严格的杂交条件下,仅仅在探针及其目标等位基因序列之间并且不在其它等位基因序列的情况下形成稳定双重物。可以通过多个众所周知的方法中的任何方法来检测稳定杂交双重物的存在。在特定实施例中,来自包括多态位点的目标等位基因的多个核酸序列被扩增并且在充分严格的杂交条件下被杂交到一组探针。根据探针至经扩增的目标序列的结合的模式来推断存在的等位基因。在该实施例中,实施扩增以便提供足够的核酸来用于通过探针杂交进行分析。因此,引物被设计使得包括多态位点的各种等位基因的区域被扩增而不管在样本中存在的等位基因。通过使用杂交到等位基因的所保存的区域的引物来实现独立于等位基因的扩增。技术人员将认识到,典型地,扩增系统的实验优化是有帮助的。可以通过使用“taqman”或“5'-核酸酶试验”来实施基于探针的基因型分型,如在以下文献中所述:专利号为5,210,015;5,487,972;和5,804,375的美国专利;以及holland等人,1988,proc.natl.acad.sci.usa,88:72767280。在taqman试验中,在扩增反应混合期间添加在经扩增的区域内杂交的经标记的检测探针。探针被修改以便防止探针充当用于dna合成的引物。通过使用dna聚合酶来实施扩增,所述dna聚合酶具备5'到3'核酸外切酶活性,例如tthdna聚合酶。在扩增的每个合成步骤期间,杂交到来自被扩展的引物下游的目标核酸的任何探针通过dna聚合酶的5'到3'核酸外切酶活性而被降级。因此,新目标链的合成还导致探针的降级,并且降级产物的积累提供目标序列合成的度量。在特定实施例中,利用两个荧光染料来标记检测探针,所述两个荧光染料中之一能够抑制(quench)其它染料的荧光性。染料被附连到探针,优选地一个附连到5'末端并且另一个附连到内部位点,使得当探针处于非杂交状态中的时候发生抑制并且使得在两个染料之间中发生通过dna聚合酶的5'到3'核酸外切酶活性对探针的分裂(cleavage)。扩增导致在抑制的相伴消除的情况下在染料之间的探针的分裂,以及在从最初被抑制的染料可观察到的荧光性中的增大。通过测量反应荧光性中的增大来监视降级产物的积累。专利号为5,491,063和5,571,673的美国专利描述了用于检测与扩增相伴地发生的探针的降级的替代方法。可以在特定于等位基因的扩增引物的情况下使用taqman试验,使得探针仅仅被用于检测经扩增的产物的存在。这样的试验被实施,如针对上述基于动力学pcr的方法所描述的那样。可替代地,可以在特定于目标的探针的情况下使用taqman试验。无论底层试验方法如何,本文中所述的系统和方法通过如下来标识样本中存在的特定序列变体:比较针对每个所分析的序列的pcr增长曲线从线性的相对偏离并且将该值与针对可存在于样本中的序列变体的所定义的阈值矩阵进行比较。图1示出了用于检测目标核酸序列中的至少一个序列变体的系统100的框图,所述系统100包括核酸扩增模块101、存储器102、处理器103和显示器104。核酸扩增模块101包括一个或多个样本处理模块105。每个样本处理模块105包括一个或多个单元或站,其用于实施对于处理包括用于分析的一个或多个核酸序列的样本所需要的各种步骤。样本处理模块105包括反应腔室106和热电冷却设备107,例如热循环器,以及可选地以下各项(未被示出)中的一个或多个:样本分发站、分离站、以及一个或多个可消耗的和/或试剂存储站。反应腔室被配置成在一个或多个核酸扩增反应步骤期间收容样本。另外,核酸扩增模块101还包括至少一个控制单元108,所述控制单元108被电连接到样本处理模块中的一个或多个。控制单元108还包括分析模块109,所述分析模块109被配置成分析核酸以获得可检测的信号。存储器102可以包括以下各项的任何组合:任何类型的易失性或非易失性存储器、诸如随机存取存储器(ram)、诸如电可擦除可编程只读存储器(eeprom)之类的只读存储器、闪速存储器、硬驱动器、固态驱动器、光盘以及诸如此类。为了简明,存储器102在图1中被描绘为单个设备,但是领会到存储器102还可以跨两个或更多设备分布。处理器103可以包括任何类型的一个或多个处理器,诸如中央处理单元(cpu)、图形处理单元(gpu)、专用信号或图像处理器、现场可编程门阵列(fpga)、张量处理单元(tpu)等等。虽然处理器103在图1中被描绘为单个设备,但是处理器103还可以跨任何数目的设备分布。可以通过使用任何合适的技术、诸如lcd、led、oled、tft、等离子体等等来实现显示器104。在一些实现方式中,显示器104可以是触敏显示器(触摸屏)。系统100还可以可操作地连接到一个或多个计算设备(未被示出),诸如台式计算机、膝上型计算机、平板设备、智能电话、服务器、专用计算设备、或能够执行本文中所述的技术和操作的任何其它(一个或多个)类型的(一个或多个)电子设备。在一些实施例中,系统的元件以及每个元件的子部件可以在单个设备中被提供或作为一起实现本中所讨论的各种功能性的两个或更多设备的组合被提供。例如,核酸扩增模块101可以包括经由一个或多个局域网和/或广域网而被通信地耦合到彼此的一个或多个服务器计算机以及一个或多个客户端计算机。最后,系统100还可以包括一个或多个外围设备110(例如打印机111和键盘112,如图1中所示),并且计算机子系统可以经由系统总线而被互连。耦合到i/o控制器的外设和输入/输出(i/o)设备可以通过本领域中已知的任何手段、诸如串行端口而被连接到系统100。例如,串行端口或外部接口(例如以太网、wi-fi等等)可以用于将系统100连接到诸如因特网的广域网、鼠标输入设备或扫描仪。经由系统总线的互连允许中央处理器103与每个子系统通信,并且控制来自系统存储器102或(一个或多个)存储设备(未被示出)的指令的执行,以及信息在子系统之间的交换。(一个或多个)系统存储器和/或存储设备可以具体化计算机可读介质。本文中所提及的任何值可以从一个部件被输出到另一部件,并且可以被输出给用户。计算机系统可以包括多个相同的部件或子系统,其例如通过外部接口或通过内部接口而被连接在一起。在一些实施例中,计算机系统、子系统或装置可以通过网络而通信。在这样的实例中,一个计算机可以被视为客户端,并且另一计算机是服务器,其中每一个都可以是相同的计算机系统的部分。客户端和服务器可以各自包括多个系统、子系统或部件。应当理解到,本公开内容的任何实施例可以如下被实现:以控制逻辑的形式,使用硬件(例如专用集成电路或现场可编程门阵列),和/或使用计算机软件,其利用以模块化或集成方式的一般可编程处理器。如本文中所使用的,处理器包括相同集成芯片上的多核处理器、或单个电路板上或联网的多个处理单元。基于本文中所提供的公开内容和教导,本领域中的普通技术人员将知道并且领会用于通过使用硬件以及硬件与软件的组合来实现本公开内容实施例的其它方式和/或方法。本申请中所描述的软件部件或功能中的任何可以被实现为将由处理器执行的、使用任何合适的计算机语言的软件代码,所述计算机语言诸如例如java、c++、或perl,其使用例如常规或面向对象的技术。软件代码可以作为一系列指令或命令被存储在用于存储和/或传输的计算机可读介质上,合适的介质包括随机存取存储器(ram)、只读存储器(rom)、诸如硬驱动器或软盘之类的磁性介质、或者诸如光盘(cd)或dvd(数字通用盘)之类的光学介质、闪速存储器以及诸如此类。计算机可读介质可以是这样的存储或传输设备的任何组合。这样的程序还可以通过使用载波信号被编码并且被传输,所述载波信号被适配用于经由符合各种协议的有线、光学和/或无线网络、包括因特网的传输。这样,可以通过使用利用这样的程序被编码的数据信号来创建根据本公开内容的实施例的计算机可读介质。被编码有程序代码的计算机可读介质可以与可兼容设备一起被封装或与其它设备分离地被提供(例如经由因特网下载)。任何这样的计算机可读介质可以驻留在单个计算机程序产品(例如硬驱动器、cd、或整个计算机系统)上或其内,并且可以存在于系统或网络内的不同的计算机程序产品上或其内。计算机系统可以包括监视器、打印机、或用于将本文中所提及的任何结果提供给用户的其它合适的显示器。本文中所述的任何方法可以完全或部分地利用计算机系统来被执行,所述计算机系统包括一个或多个处理器,其可以被配置成执行步骤。因此,实施例可以针对被配置成执行本文中所述的任何方法的步骤的计算机系统,其潜在地具有执行相应的步骤或相应的步骤组的不同部件。尽管被呈现为经编号的步骤,但是本文中的方法的步骤可以同时或以不同次序被执行。另外,这些步骤的部分可以与来自其它方法的其它步骤的部分一起使用。而且,步骤的全部或部分可以是可选的。另外,任何方法的任何步骤可以利用用于执行这些步骤的模块、电路或其它构件来被执行。图2(a)图示了由图1的系统用于检测目标核酸序列的序列变体的方法。一旦样本被接收并且被适当地处理,例如组合样本中存在的各种核酸与一个或多个引物和探针以及对于进行核酸扩增反应所需要的附加反应部件(201),样本中的核酸就经受扩增反应,并且通过使用可检测的半部、例如不同的荧光染料来标识每个核酸(202)。在特定实施例中,样本中存在至少两个核酸,例如第一变体和第二变体,并且每一个通过使用不同的可检测的半部而在扩增反应中唯一地可标识。可选地,所述方法还包括控制物质,例如管家基因,其也被唯一地标记,使得它可以在扩增反应中有区别地被检测。系统为每个扩增反应生成增长曲线(gc;203),并且然后处理器确定每个增长曲线从线性的偏离(dl;204)。从线性的偏离与彼此相比较以标识从线性比的偏离(dlr;205),即每个数据集从线性的相对偏离的比较,并且从线性比的偏离然后与特定于目标核酸序列的预定义的阈值矩阵相比较(206)以标识样本中存在的特定序列变体(207)。步骤203-207构成方法的数据分析部件(208),并且这些步骤在图2(b)中被更详细地图示。处理器接收用于第一变体(v1)的第一核酸扩增反应的第一数据集以及用于第二变体(v2)的第二核酸扩增反应的第二数据集(210)。每个数据集由处理器用于为每个扩增反应生成增长曲线(211和212),并且随后由处理器生成每个增长曲线从线性的偏离(213和214)。通过比较增长曲线从线性的偏离,处理器生成从线性比的偏离(215),其然后与特定于序列的阈值矩阵(tm)(217)相比较(216)以标识样本中存在的序列变体(218)。通过分析多个已知的特定核酸序列的单独增长曲线来确定特定于序列的阈值矩阵(217)。例如,如果将评估两个变体,则每个变体的样本池被分析以标识针对那些序列的预期增长曲线。然后,确定每个可能的感兴趣的变体相对于(一个或多个)其它的从线性比的偏离,并且该数据用于生成特定于序列的阈值矩阵。另外,阈值矩阵还可以包括增长曲线的其它特性的评估,诸如但不限于deltactr(德尔塔ctr)或deltact(德尔塔ct)。循环阈值(ct)是对于可检测的信号跨过阈值、超过背景水平、指示扩增已经开始所需要的循环的数目。ct特定于反应中存在的核酸的量。德尔塔ct对应于被评估的核酸的ct相对于参考序列的核酸的ct之间的差异。增长曲线形状可以特定于特定的感兴趣的序列、引物、探针和热循环条件。阈值矩阵包括针对在评估下的每个变体的从线性比的一组可接受的偏离的范围。例如,如在以下示例中更详细地描述的,阈值矩阵可以用于在序列中的不同等位基因、例如gg、ga和aa之间进行区分,如图3中所示。在核酸扩增模块中的三个通道中进行核酸扩增,每个通道被配置成通过使用不同的可检测的标记来检测不同的等位基因。例如,第一通道可以包括通过使用例如cy5.5所标识的控制,第二通道用于评估可检测地利用hex标记的g的存在,并且第三通道用于评估可检测地利用fam标记的a的存在。(可检测的标记的选择和特定的等位基因是非限制性的并且在本文中被提供仅仅用于说明性目的。)阈值矩阵包括针对每个等位基因从线性比的偏离的一组可接受的范围,因此对于在该示例中被评估的三个等位基因,矩阵将包括gg范围、ga范围和aa范围。在两个范围之间的接合处,可存在被称为缓冲区的范围,所述缓冲区是如下矩阵区域:在所述矩阵区域中,如果从线性比的偏离落在该区内,则可以采用一个或多个附加的计算来进一步确认等位基因的身份。如果在每个通道中的扩增之后获得经验上有效的结果(301),例如针对控制通道的增长曲线ct不在所指定的范围的外部或不存在、针对fam和hex的ct不是二者都不存在,以及针对fam和hex的单独ct不在所指定的范围的外部,于是如下评估线性比:●如果线性比大于或等于1.0(302),于是等位基因是gg;●如果线性比在-0.2与0.6之间并且存在deltact(303),于是等位基因是ga;●如果ga缓冲区中的线性比在0.6与1.0之间并且deltact在-4.0与1.0之间(304),于是等位基因是ga;●如果aa缓冲区中的线性比在-0.6与-0.2之间并且deltact大于或等于3.0或缺失(305),于是等位基因是aa;或者●如果线性比小于或等于-0.6(306),于是等位基因是aa。在特定的实施例中,所述方法包括根据在某个范围的循环数之上的核酸扩增数据来计算deltab,deltab被定义为从直线ŷ=a+bx的线性的相对最大偏离。因而,deltab可以用于计算针对核酸n的增长曲线从线性的偏离,例如deltabn=(最大|ŷi-yi|/广义基线平均数)在n对应于在被分析的核酸并且广义基线平均数包括y中位数、几何平均数、调和平均数或广义平均数的情况下,并且在特定的实施例中,广义基线平均数是y中位数。因此,deltab可以被表示如下:deltabn=(最大|ŷi-yi|/y中位数)。优选地在某个范围的循环数之上评估从线性的偏离,例如从循环m到p,其中0<m<p。在一个实施例中,m≥2,具体地,3≤m≤7,并且优选地m=6。因此,在该实施例中,从线性比的偏离,其反映表示第一和第二核酸的扩增的两个增长曲线从线性的相对近似偏离,可以被计算为:从线性比的偏离=deltab1/deltab2。在特定的实施例中,从线性比的偏离包括:log10(deltab1/deltab2)。因此,从线性比的偏离的一个示例是logdeltab比(ldbr)。令人惊讶地已经发现使用从线性比、诸如ldbr的偏离使得rfi(相对荧光强度)能够相对于常规算法被降低,并且它标识与基于与sanger定序的比较的常规方法相比具有更高灵敏度的序列变体。当已经发现具有一个或多个材料或条件的具有挑战性的样本通过使用常规逻辑而引入了基因型分型误差的时候,发现ldbr在标识正确的基因型、而不是无效或不正确的结果中是更稳健的。因而,在本文中所述的系统和方法中,通过处理器执行一种方法,包括:(a)接收第一数据集,所述第一数据集表示针对第一核酸变体扩增的增长曲线;(b)接收第二数据集,所述第二数据集表示针对附加核酸变体扩增的增长曲线,其中所述附加核酸变体包括在相对于第一核酸变体的多态位点处的所述至少一个序列修改;(c)生成拟合所述第一数据集的第一曲线以及拟合所述第二数据集的第二曲线;(d)生成第一曲线的deltab和第二曲线的deltab;(e)通过取针对第一和第二曲线的deltab的比的log10以导出ldbr,比较针对第一和第二曲线的deltab;(f)比较ldbr与阈值矩阵;以及(g)基于所述比较步骤(f)来标识所述两个或更多序列变体。特定实施例的具体细节可以用任何合适的方式来被组合。然而,本公开内容的其它实施例可以针对与每个单独方面或这些单独方面的特定组合有关的特定实施例。已经为了说明和描述的目的而呈现了本公开内容的示例性实施例的以上描述。它不意图是穷举的或将本公开内容限制到所述精确形式,并且鉴于以上教导,许多修改和变型是可能的。实施例被选择和描述以便最佳地解释本公开内容的原理及其实际应用,从而使得本领域中的其他技术人员能够在各种实施例中并且在具有如适于所设想的特定使用的各种修改的情况下最佳地利用本公开内容。示例用于基因型分型试验的算法选择和截止确定发现常规基因型逻辑在应力条件下(例如当pcr抑制剂被掺入样本中的时候)标识不正确的基因型。这些不正确的结果是由于在cobas®4800系统中的fam(具有苏氨酸连接基团的6-羧基荧光素)或hex(具有苏氨酸连接基团的六氯荧光素)通道中的rfi(相对荧光强度)值中的变化,其导致跨过rfi截止。通过使用常规方法,如果rfi值在截止以下,则为该通道移除ct值。随后,在仅仅存在于通道子集中的ct值上确定基因型(参见例如图4)。例如,如果famrfi值降落到杂合试样ga中的截止以下,将仅仅存在hex和cy5.5ct,其导致gg基因型的不正确的标识,如果所有其它值都在规范内的话。为了开发更稳健的算法,其它算法被评估。基于deltab来开发新算法,所述deltab被定义为从直线ŷ=a+bx的线性的相对最大偏离,其根据在某个范围的循环数上的实时pcr数据,如图5中所示。经改进的算法使用logdeltab比(ldbr),并且所述算法在图6中示意性地被图示。通过使用经改进的算法所获得的结果在图7中被示出,并且如表1中所图示的,新算法使得rfi能够相对于常规算法被降低。表1:rfimin-老算法相对于新算法通道老算法新算法cy5.5(ic)65fam(a等位基因)20hex(g等位基因)20ct被设置成针对具有在截止以下的rfi值的样本不存在。在通过使用cobas®x480用于样本预备的样本筛选期间,老算法和新算法二者针对>99%的试样都导致了与sanger定序相比的一致的结果(表2)。表2:老算法相对于新算法:试样筛选。相比于利用sanger定序的gg,一种试样是基因型分型不一致的ga/无效的(新/老算法),并且相比于利用sanger定序的ga,一种试样是基因型分型无效的/ga(新/老算法)。那两个试样对于其sanger基因型踪迹都是无定论的,因为它们仅仅展现了另一等位基因的一些背景,并且通常不是纯合或杂合的。另外的调研指示了那两个试样在试样收集期间可能已经被交叉污染。通过使用常规方法,这样的无定论的结果将会迫使用户重复实验,并且如果试样在试样收集期间被交叉污染,则用户可能必须收集新鲜样本来重复实验。当比较不同的样本预备与sanger定序的时候,老和新算法二者都示出了与sanger定序100%一致的结果(表3)。然而,老算法将三个试样称为无效,这是由于对于具有较高dna产出的样本预备方法,famctr值在截止范围的外部。表3:老基因型分型算法相对于新基因型分型算法:方法相关性a由于在截止范围外部的famctr值而无效。新基因型算法不使用ctr。内部控制(ic)用作针对dna品质和数量的控制。为了模拟拙劣的样本预备,运行一系列有挑战性的条件,包括pcr抑制剂的掺入、样本稀释、模拟不同洗脱缓冲液等等,并且在那些应力条件下,新算法具有相比于sanger定序的100%一致的结果,或者样本由于拙劣的dna数量或品质而无效(表4)。然而,老算法仅仅具有相比于sanger定序的97%一致的结果,并且相比于新算法,46个更多的样本(或17%)被称为无效的(表4)。表4:老基因型分型算法相对于新基因型分型算法:ic应力测试。当掺入pcr抑制剂(血液、乙醇)的时候,在老算法的情况下,19个ga样本被不正确地称为gg(表4)。这是由于famrfi值降落到rfimin截止以下,并且随后从fam通道移除ct值。因此,样本仅仅展现hex和cy5.5ct值,并且被基因型分型为gg(图8)。利用新算法,这些样本被正确地基因型分型。出于以下各种原因,在老基因型算法的情况下,更多的样本是无效的:●较高的rfimin,由于降落到截止以下的cy5.5或fam(仅仅aa)而使一些样本无效(图9),●由于δct值在范围外部而使ga试样无效(图10),或者●由于hexrfi值在截止以上而使aa试样无效。然后将样本视为ga样本,因为所有通道(fam、hex和cy5.5)ct存在,但是未通过δct截止(图11)。因此,在具有pcr抑制剂、交叉污染、高和低dna浓度的有挑战性的样本的时候,并且对于某些mastermix部件改变(数据没有在此处示出),发现使用ldbr的新基因型算法在标识正确的基因型、而不是无效或不正确的结果中是更稳健的。使用老算法导致无效的结果,其将会需要用户重复实验,这通常是不可能的。然而,新算法对否则通过使用常规方法将会被认为无效的样本进行正确的基因型分型。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1