通过快速LAMP分析进行染色体评估的制作方法
本申请要求2017年10月19日提交的美国临时申请号62/410,362的权益,将其披露以其整体并入本文。
序列表的并入
名为“lav0002201us_st25”的文件中包含的序列表以电子方式随本文一起提交并通过引用并入本文,该文件如在microsoftwindows操作系统中测量的为3.83千字节并且创建于2017年10月19日。
本文披露了确定核苷酸序列相对于其他核苷酸序列在复杂基因组中的丰度的方法。
背景技术:
染色体完整性是胚胎、胎儿和新生儿发病率和死亡率的最重要决定因素。因此,对非整倍性、拷贝数变异(cnv)和遗传性别进行测试对于通过体外受精(ivf)产生的胚胎植入前遗传筛查(pgs)、产前检查、心脏或形态异常和外生殖器性别不清的新生儿检查以及妊娠丢失后妊娠物的评价是至关重要的。
对非整倍性、cnv和遗传性别进行测试的现有方法包括g带核型分析、多重连接依赖性探针扩增(mlpa)、微阵列分析、荧光原位杂交(fish)和下一代测序(ngs)。不幸的是,这些方法需要一体化高复杂性实验室,价格昂贵,并且不能在资源贫乏的环境中进行。另外,它们需要超过24小时才能完成,从而增加了焦虑,并且可能延迟或者甚至妨碍诊断和治疗。
环介导的等温扩增(lamp)是一种相对较新的技术,其使用特异性引物和具有高链置换和复制活性的dna聚合酶,在一步过程中在等温条件下快速扩增特定dna靶标。通过将dna扩增与报告系统结合,可以检测靶序列的存在。反应在恒定温度(通常为60℃-65℃)下发生,消除了对昂贵热循环仪的需要。由于扩增反应的性质并且不需要在反应温度之间循环,因此在短至10-15分钟内快速发生扩增。由于引物用于识别六种不同的dna靶序列,因此反应非常具特异性。
因为该系统稳健、快速且成本低廉,因此已经开发了多种基于lamp的测定,用于即时检测微生物(包括病毒、细菌和真菌)、食品质量控制、肿瘤检测以及牛胚胎和法医样品的性别鉴定。本披露描述了基于定量lamp的测定(qlamp)并且展示了其在人类胚胎性别鉴定和人类临床样品非整倍性测定中的应用。
技术实现要素:
在一个实施例中,本披露描述了一种分析一种或多种核酸模板的方法,该方法包括:a)提供多重反应混合物,其包含:(i)至少一种待扩增的核酸模板,(ii)至少一种核酸靶标,(iii)用于扩增该核酸模板的引物,和(iv)用于扩增该核酸靶标的引物;b)用聚合酶共扩增该复杂模板核酸和该核酸靶标,其中该共扩增等温地发生并且包括至少94℃的热脉冲步骤,其中核酸模板和核酸靶标被扩增;c)使该核酸模板归一化,其中在完成步骤b)的共扩增后进行该归一化。
在另一个实施例中,总扩增的对照核酸的量和总核酸模板的量与该核酸模板的完整扩增相关,其中该共扩增步骤等温地进行。
在又另一个实施例中,该等温共扩增步骤包括但不限于基于核酸序列的扩增(nasba)、环介导的扩增(lamp)、解旋酶依赖性扩增(hda)、滚环扩增(rca)、重组酶聚合酶扩增(rpa)和多重置换扩增(mda)。
在另一个实施例中,本文披露的方法可以用于对非整倍性、拷贝数变异(cnv)和遗传性别进行测试。
在另一个实施例中,本文披露的方法可以用于病原体检测。
在另一个实施例中,该一种或多种核酸模板和/或该核酸靶标是人类染色体。
在又另一个实施例中,该人类染色体是1号染色体或21号染色体。
在另一个实施例中,该聚合酶浓度为约0.28至约0.56单位/μl。在另一个实施例中,对于qlamp,tm为约60℃至约65℃;并且对于巢式pcr(nest),tm为约57℃至约72℃。在另一个实施例中,该引物浓度为约25nm至约1600nm。在另一个实施例中,镁浓度为约5mm至约8mm。在另一个实施例中,该模板dna浓度小于0.1ng。在另一个实施例中,检测到靶dna浓度的1.5倍差异。在另一个实施例中,该方法进一步包含用于扩增多种核酸靶标和多种核酸模板的多种引物。在一些实施例中,f3/b3引物对的浓度为约25nm至约200nm。在另一个实施例中,lf/lb引物对的浓度为约50nm至约400nm。在另一个实施例中,bip引物对的浓度为约200nm至约1600nm。在另一个实施例中,fip引物对的浓度为约200nm至约1600nm。在另一个实施例中,在基于sybrgreen的反应中,该fip引物对浓度为约200nm至约1600nm。在另一个实施例中,该fip引物对的浓度为约100nm至约800nm。在另一个实施例中,在基于探针的反应中,该fip引物对浓度为约100nm至约800nm。在另一个实施例中,该基于探针的反应包括存在约100nm至约800nm的fipfq-fd双链体。
序列说明
seqidno:1-人类独特y染色体靶标(sry基因[id6736,ncbi])的fip引物的序列。
seqidno:2-人类独特y染色体靶标(sry基因[id6736,ncbi])的bip引物的序列。
seqidno:3-人类独特y染色体靶标(sry基因[id6736,ncbi])的f3引物的序列。
seqidno:4-人类独特y染色体靶标(sry基因[id6736,ncbi])的b3引物的序列。
seqidno:5-人类独特y染色体靶标(sry基因[id6736,ncbi])的lf引物的序列。
seqidno:6-人类独特y染色体靶标(sry基因[id6736,ncbi])的lb引物的序列。
seqidno:7-人类独特x染色体靶标(rpa4基因[id29935,ncbi])的fip引物的序列。
seqidno:8-人类独特x染色体靶标(rpa4基因[id29935,ncbi])的bip引物的序列。
seqidno:9-人类独特x染色体靶标(rpa4基因[id29935,ncbi])的f3引物的序列。
seqidno:10-人类独特x染色体靶标(rpa4基因[id29935,ncbi])的b3引物的序列。
seqidno:11-人类独特x染色体靶标(rpa4基因[id29935,ncbi])的lf引物的序列。
seqidno:12-人类独特x染色体靶标(rpa4基因[id29935,ncbi])的lb引物的序列。
seqidno:13-人类独特参考靶标(cftr基因[id1080,ncbi])的fip引物的序列。
seqidno:14-人类独特参考靶标(cftr基因[id1080,ncbi])的bip引物的序列。
seqidno:15-人类独特参考靶标(cftr基因[id1080,ncbi])的f3引物的序列。
seqidno:16-人类独特参考靶标(cftr基因[id1080,ncbi])的b3引物的序列。
seqidno:17-人类独特参考靶标(cftr基因[id1080,ncbi])的lf引物的序列。
seqidno:18-人类独特参考靶标(cftr基因[id1080,ncbi])的lb引物的序列。
seqidno:19-对人类独特y染色体靶标(sry基因)具特异性的hsry-5iabkfq-fip寡核苷酸探针的序列。
seqidno:20-对人类独特y染色体靶标(sry基因)具特异性的hsry-36-fam-fd寡核苷酸探针的序列。
seqidno:21-对人类独特x染色体靶标(rpa4基因)具特异性的hrpa4-5iabkfq-fip寡核苷酸探针的序列。
seqidno:22-对人类独特x染色体靶标(rpa4基因)具特异性的hrpa4-3joe_n-fd寡核苷酸探针的序列。
seqidno:23-对人类独特x染色体靶标(rpa4基因)具特异性的hrpa4-36-fam-fd寡核苷酸探针的序列。
seqidno:24-对人类参考靶标(cftr基因)具特异性的hcftr-5iabkfq-fip寡核苷酸探针的序列。
seqidno:25-对人类参考靶标(cftr基因)具特异性的hcftr-36-tamsp-fd寡核苷酸探针的序列。
附图说明
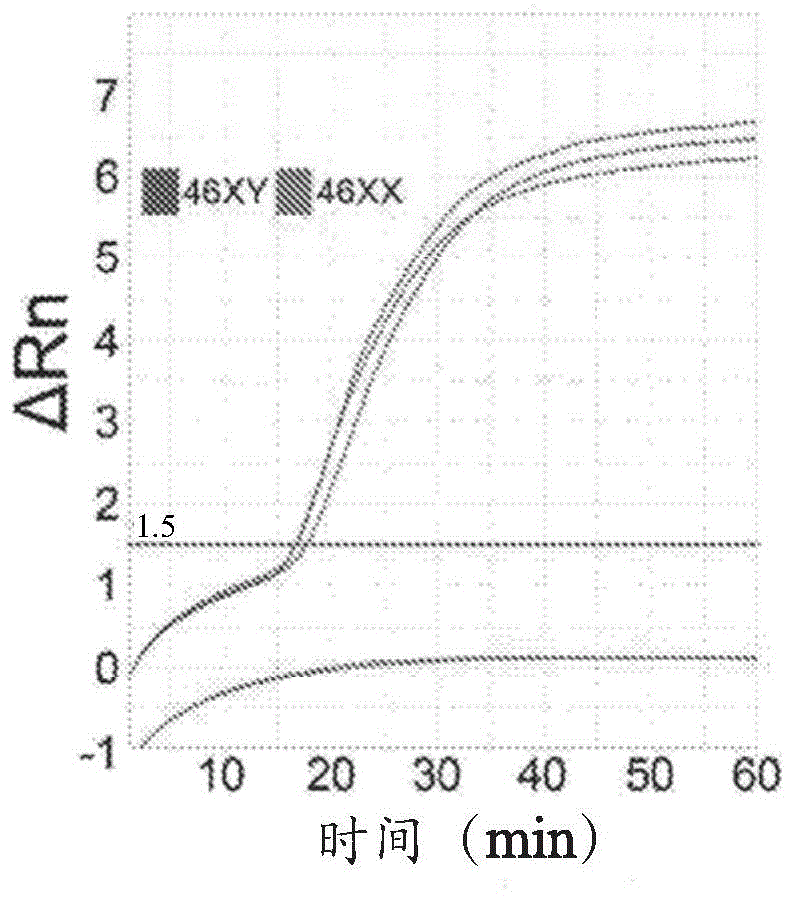
图1.图1a:在来自46xy血液的模型dna样品中lamp介导的sry靶产物(y染色体)产生的实时扩增图和在来自46xx血液的dna样品中没有扩增信号。图1b:在来自46xy和46xx血液的模型dna样品中lamp介导的rpa4靶产物(x染色体)产生的实时扩增图。图1c:在对应于不同性别核型的胚胎并用替代方法加工的af样品中lamp介导的sry靶产物(y染色体)产生的实时扩增图。af1(46xx):af1.1-冷冻/解冻的样品;af1.2-超声处理的样品。af2(46xy):af2.1-冷冻/解冻的样品;af2.2-超声处理的样品;af2.3-裂解/中和的样品。图1d:sry(y染色体)和rpa4(x染色体)靶标扩增的模型46xy对46xx的比较。
图2a:在分离的sry靶标的2倍系列稀释液中lamp介导的sry靶产物(y染色体)产生的实时扩增图。图2b:在分离的sry靶标的2倍系列稀释液中基于ct值(例如,检测相应的阳性扩增信号(阈值δrn)的时间)绘制的稀释曲线。一式三份地分析每个点的稀释液。
图3a至图3d.在从血细胞分离的人类基因组dna样品(46xy)的1.5倍系列稀释液中的全基因组内容物内的完整sry靶标(y染色体)的基于定量lamp的检测。在原始的或预热的人类基因组dna(46xy)的1.5倍系列稀释液中基于ct值(例如,检测相应的阳性扩增信号(阈值δrn)的时间)绘制的稀释曲线。图3a:原始的(未处理的)dna。图3b:在100℃下预热60sec的基因组dna。图3c:在100℃下预热90sec的基因组dna。图3d:针对原始的和预热的基因组dna的1.5倍系列稀释液绘制的稀释曲线的比较。一式三份地分析每个点的稀释液。
图4a至图4b.在从原代血细胞分离的不同性别核型(46xy对46xx)的人类基因组dna样品的2倍系列稀释液中完整rpa4靶标(x染色体)拷贝数比率的基于lamp的定量。图4a:在人类基因组dna(46xy对46xx)的2倍系列稀释液中基于ct值(例如,检测靶标扩增的相应的阳性信号(阈值δrn)的时间)绘制的稀释曲线。图4b:在从原代血细胞分离的dna样品(46xy(相对于其归一化的参考样品)对46xx)的2倍系列稀释液中rpa4靶标(例如,x染色体)的计算的和归一化的拷贝数比率的比较。一式三份地分析每个点的稀释液,并且呈现的数据是两次独立实验的平均结果。
图5.从10倍系列样品稀释液中的基因组内容物分离出的预扩增sry靶标(y染色体)的基于定量lamp的检测。图5a:示出了在分离的sry靶标的10倍系列稀释液中lamp介导的sry靶产物(y染色体)产生的实时扩增图。图5b:示出了在分离的sry靶标的10倍系列稀释液中基于ct值(例如,检测相应的阳性扩增信号(阈值δrn)的时间)绘制的稀释曲线。一式三份地分析每个点的稀释液。
图6.在不存在(正方形)和存在(圆形)无sry靶标的46xx基因组dna的情况下从2倍系列样品稀释液中的基因组内容物分离出的预扩增sry靶标(y染色体)的基于定量lamp的检测。在分离的sry靶标的2倍系列稀释液中基于ct值绘制的稀释曲线。一式三份地分析每个点的稀释液。
图7.从1.5倍系列样品稀释液中的基因组内容物分离出的预扩增sry靶标(y染色体)的基于定量lamp的检测。在分离的sry靶标的1.5倍系列稀释液中基于ct值绘制的稀释曲线。一式三份地分析每个点的稀释液。
图8.在原始未处理的和酶消化的基因组46xydna的2倍系列稀释液中lamp介导的sry靶产物(y染色体)产生的实时扩增图。图8a:原始未处理的dna;图8b:消化10min的基因组dna;图8c:消化20min的基因组dna。一式三份地分析每个点的稀释液。
图9.在原始未处理的和机械消化的(针剪切)基因组46xydna的2倍系列稀释液中的sry靶标(y染色体)的lamp导致质量差的定量。图9a:原始未处理的dna的2倍稀释液的实时扩增图。图9b:原始未处理的dna的2倍稀释液的基于ct值绘制的稀释曲线。图9c:机械消化的dna(通过来回通过胰岛素针20次剪切)的2倍稀释液的实时扩增图。图9d:机械消化的dna(通过来回通过胰岛素针20次剪切)的2倍稀释液的基于ct值绘制的稀释曲线。一式三份地分析每个点的稀释液。
图10.在从原代血细胞和培养的细胞分离的不同性别核型(46xy对46xx)的预热的人类基因组dna样品的2倍系列稀释液中完整rpa4靶标(x染色体)拷贝数比率的基于lamp的定量。在不同来源(血细胞或培养的细胞)的dna的2倍系列稀释点基于ct值(例如,检测靶标扩增的相应的阳性信号(阈值δrn)的时间)绘制的稀释曲线的比较。一式三份地分析每个点的稀释液。
具体实施方式
本披露提供了分析一种或多种核酸模板的方法,该方法包括:a)提供多重反应混合物,其包含:(i)至少一种待扩增的核酸模板,(ii)至少一种核酸靶标,(iii)用于扩增核酸模板的引物,和(iv)用于扩增核酸靶标的引物;b)用聚合酶共扩增复杂模板核酸和核酸靶标,其中共扩增等温地发生并且包括至少94℃的热脉冲步骤,其中核酸模板和核酸靶标被扩增;c)使核酸模板归一化,其中在完成步骤b)的共扩增后进行归一化。
对遗传异常进行快速、负担得起、即时的测试一直是植入前、产前诊断和产后诊断中分子诊断的长期目标。用于染色体评估的现有方法耗时、昂贵,并且需要一体化高复杂性实验室。因此,延误了诊断和治疗,并且在资源贫乏的环境中测试通常不是一种选择。
本发明人的目标是开发一种快速、廉价的测试,其可以即时地进行以检测染色体非整倍性。在这里,改编先前用于靶dna序列的定性分析的定量环介导的等温扩增(qlamp)以用于定量检测染色体非整倍性。优化了报告基因测定,并且使用靶标特异性荧光探针,其使得能够实时监测产物扩增期间的荧光增加。然后使用六个引物对测试外周血、细胞培养物和羊水样品的两个独特的单拷贝靶基因:分别是染色体y和x上的sry和rpa4基因。
在这些条件下,使用少至1ng的基因组dna,可以容易地区分雄性和雌性样品,并且在20分钟内鉴定46xx、46xy、47xyy和45x0核型。因此,qlamp可以在20分钟内评估基因组dna靶标的相对拷贝数,而无需复杂的实验室设备或训练。当优化和改编时,此测定可以使得能够准确筛选所有染色体的cnv和非整倍性。
根据本披露,核酸模板可以是dna或rna。例如,在一些实施例中,核酸模板可以是选自下组的核酸,该组由以下组成:cdna(互补dna)、lna(锁核酸)、mrna(信使rna)、mtrna(线粒体rna)、rrna(核糖体rna)、trna(转移rna)、nrna(核rna)、dsrna(双链rna)、核酶、核糖开关、病毒rna、dsdna(双链dna)、ssdna(单链dna)、质粒dna、粘粒dna、染色体dna、病毒dna、mtdna(线粒体dna)、ndna(核dna)。在一些实施例中,核酸模板可以是多于一种此类核酸类型。此外,核酸模板也可以是一组核酸的全部,优选分别是mrna或cdna的全部(转录组),和/或一种或多种生物的dna的全部(基因组)。本领域技术人员知道rna的全部可以代表生物的基因组,这尤其可以适用于rna病毒。如本文所述的核酸模板也可以是一个或多个染色体。
待分析的核酸模板可以具有不同的来源。例如,核酸模板可以从选自下组的一种或多种生物中分离,该组由以下组成:病毒、噬菌体、细菌、真核生物、植物、真菌和动物。在一些实施例中,核酸模板可以来自特定细胞类型,如包括但不限于血细胞或免疫细胞(如b或t淋巴细胞)。其他实施例提供了分离或源自任何细胞系(如成纤维细胞系)的样品。可以使用任何特定的细胞类型或细胞系。此外,待分析的核酸模板可以是特定样品的一部分或一部份。此类样品也可以具有各种来源。因此,本披露的方法还可以涉及对包含在例如环境样品(如水样品)中的核酸的分析。在一些实施例中,设想样品可以获得自可以从其获得核酸模板的任何来源,包括但不限于生物样品,如血液样品、血清样品、羊水样品、脑脊液样品等。
如本文所用,等温反应旨在指代仅在一个温度下进行的反应。如果在反应开始之前(例如,在冰上或在冷藏条件下孵育、储存或运输)或在反应完成之后(例如,以便灭活反应组分或酶)反应温度改变,则反应仍被称为等温的,只要反应本身在恒定温度下进行即可。如果温度变化不超过+/-10℃,则认为温度是恒定的。
i.染色体特异性靶标和lamp引物设计。
通过挖掘文献和数据库(国际hapmap项目,entrezsnp,decipher(使用ensembl资源的人类染色体不平衡和表型数据库(databaseofchromosomalimbalanceandphenotypeinhumansusingensemblresources),decipherconsortium))为每个人类性染色体鉴定和选择两个具有不变基因座的独特单拷贝靶基因:对y染色体具特异性的sry(性别决定区y[id6736,ncbi])和对x染色体具特异性的rpa4(复制蛋白a4[id29935,ncbi])。在lamp引物设计软件primerexplorerv4的帮助下为每个靶基因设计六种引物(fip、bip、f3、b3、lf、lb)的组合的lamp引物组。引物设计和优化在本领域是熟知的。尽管如本文所述的引物用于展示本披露的方法,但是这些方法不旨在限于特定的引物序列。本领域技术人员将认识到,在不背离本披露的范围的情况下,可以使用具有类似成功结果的其他软件程序和/或引物序列。
表1.用于人类独特y染色体和x染色体靶基因以及参考靶基因(cftr)的lamp引物。
用作信号归一化的内部参考对照的cftr(囊性纤维化跨膜传导调节因子[id1080,ncbi])基因靶标的lamp引物已在其他地方公开。
设计并合成了由荧光标记的fip引物代表的另外的探针,该引物与和猝灭剂连接的互补fd寡核苷酸一起对齐,以确保基于荧光的靶标特异性读出,以在单重/多重测定中检测和区分每种独特的靶标。引物序列提供在表2中。
表2.用对人类独特y染色体和x染色体靶基因和参考靶基因(cftr)具特异性的荧光染料或猝灭剂标记的寡核苷酸-探针对。
ii.靶dna样品
对于qlamp的初步开发,将来自人类sry基因(性别决定区y[id6736,ncbi])和rpa4基因(复制蛋白a4[id29935,ncbi])的200-bppcr产物用作靶序列,该产物使用qiaquickpcr纯化试剂盒(凯杰公司(qiagen))纯化,随后通过sanger进行确认。所使用的引物序列提供在表2中。根据本披露可以使用任何独特的靶基因,只要所选择的靶基因为特定应用提供特异性鉴定即可。单拷贝数基因可以提供有益的结果,或者也可以使用足够低拷贝数的基因,这取决于特定的用途。如本文所述,可以使用任何浓度的靶基因。在一些实施例中,低浓度的靶基因核酸和/或其扩增产物在存在用于分析的最小核酸样品的情况下可能是有益的。可以根据需要进行系列稀释以提供所需浓度的靶核酸序列。例如,对于目前描述的sry和rpa4分析,首先测试纯化的pcr片段的系列稀释液以优化lamp参数以进行正确定量并表征测试限制,如本文所述。
对人类基因组样品的后续测试利用从成人原代血细胞和来自具有正常雄性(46xy)[gm12877,coriell]和雌性(46xx)[gm12878coriell]核型的b淋巴细胞的良好表征的类淋巴母细胞系分离的dna样品。性染色体比率异常(47xyy、45x0)的样品源自coriell细胞库(coriellcellrepository)的相应细胞系(例如,成纤维细胞系,xyy综合征[gm11337])和原始人体组织[指定何种类型的样品]样品。通过适当的基于柱的制造方案(
如本领域技术人员所理解的,核酸靶标或模板的扩增可能需要制备样品,例如以灭活特定的酶或蛋白质、降解特定的核酸或从基因组dna中释放二级结构。用于这种样品制备的任何特定方法都可以与本发明方法一起使用,如在特定温度下孵育样品或添加某些洗涤剂或试剂。对于本情况,将样品在100℃孵育范围从30-120sec的不同时间。未处理的完整基因组dna(对照)和预热的dna均用于制备10倍、2倍和1.5倍系列稀释液,并且使用lamp对这些dna进行测试以用于特异性靶标定量,如本文所述。
iii.lamp反应
在一些实施例中,可以使用靶标特异性荧光探针(例如,5’-猝灭剂-fip:fd-荧光团-3’(q-fip:fd-fluo)双链体)进行如本文所述的lamp分析,以使得能够特异性且敏感地检测靶基因。这种靶标特异性探针可以例如在使用前立即通过混合等体积的200μm5’-猝灭剂-fip(5’-修饰)和200μmfd-荧光团(3’-修饰)以及两倍体积的无核酸酶水以达到所需浓度来制备。根据本披露,任何特定浓度的如本文所述的靶标特异性探针和/或寡核苷酸都是有用的,如包括但不限于0.1μm、0.2μm、0.3μm、0.4μm、0.5μm、0.6μm、0.7μm、0.8μm、0.9μm、1μm、2μm、3μm、4μm、5μm、6μm、7μm、8μm、9μm、10μm、20μm、30μm、40μm、50μm、60μm、70μm、80μm、90μm、100μm、150μm、200μm、250μm、300μm等。在一些实施例中,50μm浓度的每种寡核可以提供有益的结果。本领域技术人员将认识到,可以根据需要优化靶标特异性探针和/或寡核苷酸的浓度以获得所需结果。在本情况下,将混合物在98℃下加热3min以使互补序列对齐,随后缓慢逐渐冷却至室温。
完整的lamp反应或与一种或多种靶标特异性荧光探针的反应混合物可以含有任何所需浓度的每种组分和/或试剂,以实现所需结果。为此目的,可以单独地或分开地优化此类单独组分。例如,如本文和实例中详细描述的,本披露的lamp反应可以含有例如0.8μmfip和0.4μmq-fip:fd-fluo双链体、1.6μmbip、f3和b3各0.2μm、lf和lb各0.4μm;2.8ubst2.0warmstartdna聚合酶(新英格兰生物实验室(newenglandbiolabs,neb));dntp各1mm(neb);dna模板(测试的稀释液);另外补充有6mmmgso4(neb)、1xrox参考染料(0.5μm)的1x等温扩增缓冲液。对于多重反应,也可以根据需要优化单独测定的总引物浓度。对于多重测定或反应,可以保持试剂浓度用于单次测定,或者可以改变试剂浓度以适合特定应用。本披露旨在涵盖可以提供所需结果的任何特定浓度。在一些实施例中,可以如本文所述使用试剂浓度用于标准lamp反应,其中每组引物和/或探针代表总数的1/n,其中n是在特定分析中评价的靶标和相应引物组的数目。
在一些实施例中,如本文所述的用于lamp测定的试剂浓度可以是适合于特定用途的任何浓度。例如,可以在本披露的lamp测定中使用的浓度的非限制性实例可以包括但不限于从约0.28至约0.56单位/μl的聚合酶浓度。聚合酶可以是能够实现所需结果所需的扩增的任何聚合酶。在另一个实施例中,tm为约60℃至约65℃。在另一个实施例中,引物浓度为约25nm至约1600nm。在另一个实施例中,镁浓度为约5mm至约8mm。在另一个实施例中,模板dna浓度小于0.1ng。在另一个实施例中,检测到靶dna浓度的1.5倍差异。在另一个实施例中,该方法进一步包含用于扩增多种核酸靶标和多种核酸模板的多种引物。在一些实施例中,f3/b3引物对的浓度为约25nm至约200nm。在另一个实施例中,lf/lb引物对的浓度为约50nm至约400nm。在另一个实施例中,bip引物对的浓度为约200nm至约1600nm。在另一个实施例中,fip引物对的浓度为约200nm至约1600nm。在另一个实施例中,在基于sybrgreen的反应中,fip引物对浓度为约200nm至约1600nm。在另一个实施例中,fip引物对的浓度为约100nm至约800nm。在另一个实施例中,在基于探针的反应中,fip引物对浓度为约100nm至约800nm。在另一个实施例中,基于探针的反应包括存在约100nm至约800nm的fipfq-fd双链体。
lamp反应可以在任何反应体积中进行,例如包括但不限于5μl、10μl、15μl、20μl、25μl、30μl、35μl、40μl、45μl、50μl、60μl、70μl、80μl、90μl、100μl、125μl、150μl、175μl、200μl、250μl、300μl、350μl、400μl、450μl、500μl、1ml等。在一些特定的应用中,在实验内(测定内)和在独立实验中(测定间)一式两份地或一式三份地进行反应可能是有益的。对于分析,可以将反应板在任何所需温度下或在适合于特定应用的任何温度范围下孵育。在本情况下,将反应板在62℃下孵育60min,并且使用qpcr实时循环仪(steponeplustm实时pcr系统,应用生物系统公司(appliedbiosystems))以30-sec循环步骤以实时模式进行监测。
iv.数据分析,统计。
如本文所述,可以使用任何适用的统计方法(如ct值)来执行数据分析,以便反映达到阳性信号阈值所花费的时间。这些值可以用于绘制随参考样品(46xy核型)中每种单独靶标的靶标拷贝数输入负载变化的校准曲线。测试样品的每种稀释液的相应扩增ct值可以用于计算那些样品中的输入靶标拷贝数。可以相对于参考样品稀释液的相应值归一化测试样品的每种稀释液中所获得的靶标拷贝数,以便计算靶标拷贝数比率。
可以通过学生t检验评价同时发生率的均值和方差的显著性,并且确定效应量以及用于一式两份重复和一式三份重复的每个实验内(测定内)以及独立实验之间(测定间)的p值和标准差。
v.核酸的修饰
在一些实施例中,修饰或合成核酸靶标或模板序列或分子可能是有用的。可以使用本领域技术人员熟知的许多方法来分离和操作dna分子。例如,pcr技术可以用于扩增特定的起始dna分子和/或产生起始dna分子的变体。也可以通过本领域已知的任何技术(包括通过化学手段直接合成片段)获得dna分子或其片段。因此,可以合成如本文所述的全部或部分核酸。
如本文所用,术语“互补核酸”是指能够彼此特异性杂交的两种核酸分子,其中这两种分子能够形成反平行的双链核酸结构。在这方面,核酸分子被说成是另一种核酸分子的互补序列,如果它们表现出完全互补性的话。两种分子被说成是“最小互补的”,如果它们能够以足够的稳定性彼此杂交以允许它们在至少常规的“低严格”条件下保持彼此退火的话。类似地,这些分子被说成是互补的,如果它们能够以足够的稳定性彼此杂交以允许它们在常规的“高严格”条件下保持彼此退火的话。严格条件在本领域是已知的,并且将由阅读本披露的技术人员所理解。本领域技术人员还将理解,可以视情况改变严格性以确保最佳结果。如本文所述的互补性还指代dna编辑酶与其靶标在体内或在体外的结合。本领域技术人员将认识到,互补性的变化将取决于特定的核酸序列,并且能够视情况修改条件以解决这个问题。
本披露的多核苷酸可以由rna或dna构成。优选地,多核苷酸由dna构成。主题披露还涵盖那些在序列上与本文披露的多核苷酸互补的多核苷酸。本披露的多核苷酸和多肽能够以纯化或分离的形式提供。
由于遗传密码的简并性,多个不同的多核苷酸序列可以编码本披露的多肽。在lewin(1985)中描述了显示所有可能的三联体密码子(并且其中u也代表t)和由每个密码子编码的氨基酸的表。另外,产生编码本披露的相同或基本上相同的多肽的替代多核苷酸序列完全在本领域技术人员的技术之内。这些变体或替代多核苷酸序列在本披露的范围内。如本文所用,对“基本上相同的”序列的提及是指编码不实质上改变由本披露的多核苷酸编码的多肽的功能活性的氨基酸取代、缺失、添加或插入的序列。
如本文所用,术语“序列同一性”、“序列相似性”或“同源性”用于描述两个或更多个核苷酸序列之间的序列关系。通过在特定数目的核苷酸上比较两个最佳比对的序列来确定两个序列之间的“序列同一性”百分比,其中比较窗口中序列的部分可以包含与参考序列相比的添加或缺失(即,空位)。两个序列被说成是相同的,如果每个位置的核苷酸都相同的话。在5’至3’方向上观察时的核苷酸序列被说成是在3’至5’方向上观察的第二核苷酸序列的“互补序列”或与其互补,如果第一核苷酸序列与第二序列或参考序列表现出完全互补性的话。如本文所用,当从5’至3’读取的序列之一的每个核苷酸都与当从3’至5’读取时的另一个序列的每个核苷酸互补时,则核酸序列分子被说成表现出“完全互补性”。与参考核苷酸序列互补的核苷酸序列将表现出与参考核苷酸序列的反向互补序列相同的序列。
如本文所述的核酸靶分子和核酸模板分子可以指代可以依据与本文具体例示的本披露的那些序列的同一性和/或相似性范围来定义的多核苷酸。序列同一性通常大于60%,优选大于75%,更优选大于80%,甚至更优选大于90%,并且可以大于95%。序列与本文例示的序列相比的同一性和/或相似性可以是49%、50%、51%、52%、53%、54%、55%、56%、57%、58%、59%、60%、61%、62%、63%、64%、65%、66%、67%、68%、69%、70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%。除非另有规定,否则如本文所用,可以使用karlin和altschul(1990)(如在karlin和altschul(1993)中所修改)的算法来确定两个序列的百分比序列同一性和/或相似性。这种算法被并入altschul等人(1990)的nblast和xblast程序中。可以用nblast程序(得分=100,字长=12)来执行blast检索,以获得具有所需百分比序列同一性的序列。为了获得用于比较目标的有空位的比对,可以使用如altschul等人(1997)中所述的gappedblast。当利用blast和gappedblast程序时,可以使用相应程序(nblast和xblast)的默认参数。参见ncbi/nih网站。
如本文所用,术语“核酸”和“多核苷酸”是指单链或双链形式的脱氧核糖核苷酸、核糖核苷酸或混合的脱氧核糖核苷酸和核糖核苷酸聚合物,并且除非另有限制,否则将涵盖天然核苷酸的能够以与天然存在的核苷酸类似的方式起作用的已知类似物。多核苷酸序列包括转录成rna的dna链序列和与转录的dna链互补的链序列。多核苷酸序列还包括全长序列以及衍生自全长序列的较短序列。多核苷酸序列包括作为单独链或在双链体中的有义链和反义链。
vi.试剂盒
本披露进一步提供了包含一次性容器的试剂盒,该容器包含用于进行如本文所述的lamp分析的一种或多种组分。试剂盒可以进一步包含用于核酸分离、pcr或根据披露可以有用的其他方法的试剂。
本披露的试剂盒中提供的组分可以包括例如可用于进行如本文所述方法的任何起始材料。这种试剂盒可以包含用于在多种测定(包括例如核酸测定(例如,pcr或rt-pcr测定)、遗传互补测定或根据本披露有用的任何测定)中使用的一种或多种此类试剂或组分。组分可以视情况以冻干、脱水或干燥的形式提供,或者可以在水溶液或适合根据本披露使用的其他液体介质中提供。
可用于本披露的试剂盒还可以包括另外的试剂,例如缓冲液、培养基组分(如包括mgcl2在内的盐、聚合酶、引物和脱氧核糖核苷酸等)、用于dna分离的试剂等,如本文所述。此类试剂或组分在本领域是熟知的。在适当的情况下,这种试剂盒所包括的试剂可以在与引物对或多个引物对相同的容器或培养基中提供,或者可以替代地放置在第二或另外的不同容器中,向该容器中可以放置和适当地等分另外的组合物或试剂。可替代地,能够以单个容器手段提供试剂。本披露的试剂盒还可以包括使用说明书,包括适当的单独组分的存储要求。
vii.定义
所提供的定义和方法定义了本披露,并且在本披露的实践中指导本领域普通技术人员。除非另有说明,否则术语应根据相关领域普通技术人员的常规用法来理解。分子生物学中常见术语的定义也可以在以下文献中找到:alberts等人,molecularbiologyofthecell[细胞分子生物学],第5版,加兰科学出版公司(garlandsciencepublishing,inc.):纽约,2007;rieger等人,glossaryofgenetics:classicalandmolecular[遗传学术语:经典与分子],第5版,施普林格出版社(springer-verlag):纽约,1991;king等人,adictionaryofgenetics[遗传学词典],第6版,牛津大学出版社(oxforduniversitypress):纽约,2002;和lewin,genesix[基因ix],牛津大学出版社(oxforduniversitypress):纽约,2007。使用如37cfr§1.822中阐述的dna碱基命名法。
在一些实施例中,用于描述和要求保护本披露的某些实施例的表示成分的量、性质如分子量、反应条件等的数字应理解为在一些情况下通过术语“约”修饰。在一些实施例中,术语“约”用于指示值包括用于确定该值的装置或方法的均值的标准差。在一些实施例中,书面描述和所附权利要求书中阐述的数值参数是近似值,其可以根据特定实施例寻求获得的所需性质而变化。在一些实施例中,数值参数应该根据报告的有效数字的数目并通过应用普通的舍入技术来解释。尽管阐述本披露的一些实施例的宽范围的数值范围和参数是近似值,但是具体实施例中阐述的数值尽可能精确地报告。在本披露的一些实施例中呈现的数值可能含有必然由在它们相应的测试测量中发现的标准差导致的某些误差。本文对值范围的描述仅旨在用作单独提及落入该范围内的每个单独值的简写方法。除非本文另有指示,否则每个单独值被并入说明书中,如同其在本文中单独描述一样。
在一些实施例中,在描述特定实施例的背景下(特别是在某些以下权利要求的背景下)使用的术语“一个”和“一种”以及“该”和类似提及可以被解释为涵盖单数和复数,除非另有特别说明。在一些实施例中,如本文(包括权利要求书)所用的术语“或”用于表示“和/或”,除非明确指出仅指代替代物或替代物是相互排斥的。
术语“包含”、“具有”和“包括”是开放式连接动词。一个或多个这些动词的任何形式或时态(如“包含(comprises)”、“包含(comprising)”、“具有(has)”、“具有(having)”、“包括(includes)”和“包括(including)”)也是开放式的。例如,“包括(comprises)”、“具有”或“包括(includes)”一个或多个步骤的任何方法不限于仅具有那些一个或多个步骤,并且还可以覆盖其他未列出的步骤。类似地,“包含”、“具有”或“包括”一个或多个特征的任何组合物或装置不限于仅具有那些一个或多个特征,并且可以覆盖其他未列出的特征。
本文描述的所有方法都能够以任何合适的顺序进行,除非本文另有指示或另外与上下文明显矛盾。相对于本文的某些实施例提供的任何和所有实例或示例性语言(例如,“如”)的使用仅旨在更好地阐明本披露并且不对本披露的范围构成限制,除非另有声明。本说明书中的任何语言都不应该被解释为指示任何未要求保护的要素为实践本披露所必需。
本文披露的本披露的替代要素或实施例的分组不应该被解释为限制。每个组成员可以单独地提及和要求保护,或者与该组的其他成员或本文中找到的其他元素以任何组合提及和要求保护。出于方便或可专利性的原因,组的一个或多个成员可以包括在组中或从其中删除。
已经详细描述了本披露,清楚的是在不背离所附权利要求书限定的本披露的范围的情况下,修改、变化和等效实施例是可能的。此外,应该了解,本披露中的所有实例都作为非限制性实例提供。
实例
本披露的实施例的实例提供在以下实例中。以下实例仅以说明的方式呈现,并且帮助本领域普通技术人员使用本披露。这些实例并不旨在以任何方式另外限制本披露的范围。
实例1
人类样品中染色体的定性检测
为了确定lamp是否可以定性地检测人类全基因组dna样品中的靶序列,从snp微阵列确认的正常雄性(46xy)和雌性(46xx)的外周血、成纤维细胞系以及羊水(af)样品中分离纯化的基因组dna。使用基于浊度/吸光度和荧光的报告基因测定以及先前采用和描述的检测原理进行lamp,其使用具有不变基因座的对y染色体(sry)和x染色体(rpa4)具特异性的两个独特的单拷贝靶基因的六种探针的引物组。通过实时监测产物扩增期间的荧光增加,使用靶标特异性荧光标签(探针)获得最高质量、灵敏度、特异性和准确性(图1)。在这些条件下,可以仅在15-30min内并且需要少至1-2ng的基因组dna容易地区分具有明显差别的雄性和雌性样品(图1a、图1b和图1d)。
假如原始的或处理的临床af样品含有非常低的dna浓度(在小的测试样品等分试样中数十到几个胎儿基因组拷贝),则在45-60min实现性别鉴别,这取决于样品加工方法(图1c)。
总之,这些初步结果指示,通过基于lamp的反应,可以在对照dna和af样品的存在/不存在分析中快速可靠地定性地测定每种人类dna靶标。尽管这可以作为快速且精确的胚胎和新生儿性别决定的新的有效且方便的工具,如在外生殖器性别不清的情况下,但是有用得多的是可以区分基因组dna样品中不同靶序列的相对拷贝数的定量方法。
实例2
定量靶标检测:分离的靶标
为了开发一种使用qlamp定量分析靶dna的方法,测试了qlamp检测不同浓度的纯化的小dna靶标的能力。从基因组dna模板预扩增对人类x和y性染色体具特异性的两个独特的单拷贝靶序列(分别是rpa4区和sry区)并且用qlamp进行测试。三体性(如21三体(又名唐氏综合症))代表受影响染色体数目增加1.5倍。因此,为了成为有效的筛选或诊断测试,qlamp需要检测靶dna浓度的1.5倍差异。我们制备了范围从1.5x1011至150个拷贝的分离的sry靶标的10倍(图5)、2倍和1.5倍系列稀释液。在整个稀释范围内,10x稀释液的qlamp校准曲线是高度可再现的拷贝(图2a、图2b;图6)。对于2倍和1.5倍稀释液,分别在1.5x105-1172(最大sd=0.2278)和2.6x104-3424(sd=0.1999)sry靶标拷贝范围内获得了高度可再现的结果(图7)。
为了确定基因组dna的存在是否干扰qlamp,在存在等摩尔基因组拷贝数的缺少sry靶序列的46xx基因组dna的情况下测试系列稀释液。
如图6所示,46xx基因组dna的存在不干扰扩增的准确性和效率,并且仅在扩增过程中引起轻微延迟。总之,这些数据指示,至少在分离的dna水平上,即使初始输入小至1172靶标拷贝数,也可以容易且可靠地分辨靶标拷贝数的2倍至1.5倍差异。还确认了先前公开的观察结果,即非靶dna本身的存在似乎不影响检测的准确性和精确度。
实例3
定量靶标检测:全基因组样品
接下来,测试了lamp在从外周血样品和细胞培养物中分离的全基因组dna内定量地测量靶标浓度的浓度差异的能力。首先评价了基于dna质量和片段化等温扩增(lamp)和定量的靶标可及性如何改变。通过酶消化(非位点特异性核酸酶,如片段酶或微球菌核酸酶)、机械消化(剪切或超声处理以部分消化dna)和dna预热来处理基因组dna样品。用qlamp测试所得样品以优化dna样品加工的其他步骤。显示靶基因组dna的酶消化和机械消化不足并且不能提高qlamp效率、准确性、精确度以正确定量靶序列(图8和图9)。取决于消化的时间和强度,在重复内检测的扩增率和可再现性不足以绘制稀释曲线并检测片段化输入dna的系列稀释液之间的差异(图8),或者至少与原始的(未处理的)dna的扩增率和可再现性相当(图9)。推测起来,这些处理程序引起dna损伤,这损害了qlamp有效扩增靶序列的能力。
据假设,短暂的热变性步骤可能会从人类基因组dna样品中去除三级dna结构,从而提高lamp的靶标可及性并减少样品之间的差异,从而提高测定的准确性和精确度。将基因组dna样品预热至100℃,时间间隔范围从10-180sec,将其扩增率与原始基因组dna和分离的靶标进行比较。如图3所示,预热加热基因组dna样品提高了sry靶标扩增效率、可再现性和准确性(图3)。
实例4
性染色体的定量
接下来在源自血细胞和细胞培养物的正常雄性和雌性核型(46xy、46xx)的全基因组dna样品中评估lamp测量相对x染色体含量的能力。目的是可靠地区分x染色体拷贝数的2倍比率。重复地制备每种预热dna样品的范围从85.8ng(26x103个基因组拷贝)至10.7ng(3250个基因组拷贝)的相同的2倍系列稀释液,并且用基于lamp的测定进行测试以定量相对rpa4靶标拷贝数。将对应于达到阳性信号的扩增时间的所获得的ct值用于绘制稀释曲线(图4a)。将46xy样品用作信号归一化的参考样品。根据描述参考46xy的稀释(标准)曲线的等式计算每个稀释点的46xx样品中的rpa4靶标拷贝数(图4a)。在相应的稀释点,相对于46xy样品中的参考拷贝数归一化在46xx样品中检测到的rpa4靶标(即,x染色体)拷贝的计算的数目。计算每个稀释点的一式三份重复的标准差(测定内),并且也相对于参考样品进行归一化。在两次独立实验中重复和再现该测试(测定间);也计算了测定间标准差(图4a和图4b)。因此,lamp可以测量使用从外周血样品中分离的预热基因组dna样品区分46xy和46xx核型的基因组dna样品所需的rpa4的预期1:2相对定量。相比之下,即使在预热步骤之后,源自培养的细胞系的dna仍显示lamp质量差,无法正确靠近和定量rpa4靶标(图10)。每个稀释点的重复信号偏差都特别高,大大降低了分析的准确性。
总的来说,数据证明了lamp定性地检测从外周血、组织和羊水中提取的人类基因组dna样品中的靶序列,更重要的是定量地测量靶序列的相对水平(例如,染色体拷贝数)的有利能力。
实例5
lamp的优化
如下所述进行能够检测1:2差异的定量2重lamp。特别地,lamp实验中使用的酶浓度增加,tm增加,并且引物浓度降低。另外,对于液滴测定实验,mg2+浓度降低,导致比用超低dna输入<0.1ng的定量lamp实验更稳健且具特异性的反应。
也进行了双重lamp以用较低dna输入进行非整倍性检测。例如,使用不同浓度(包括60ng、30ng、10ng和3ng)的dna面板(panel)(46xx、46xy、45xo、47xxx)。
通过引入更多技术重复,即(3个对10个重复)以及另外的引物对,提高了检测灵敏度和能力。
实例6
染色体特异性靶标和lamp引物设计。
使用等温pcr进行非整倍性检测的进一步测试。通过挖掘文献(15)和数据库(国际hapmap项目,entrezsnp,decipher(使用ensembl资源的人类染色体不平衡和表型数据库(databaseofchromosomalimbalanceandphenotypeinhumansusingensemblresources),decipherconsortium))(16,17)为每个人类性染色体鉴定和选择两个具有不变基因座的独特单拷贝靶基因:对y染色体具特异性的sry(性别决定区y[id6736,ncbi])和对x染色体具特异性的rpa4(复制蛋白a4[id29935,ncbi])。在lamp引物设计软件primerexplorerv4的帮助下为每种靶标设计六种引物(fip、bip、f3、b3、lf、lb)的lamp引物组(si,表1)。用作信号归一化的内部参考对照的cftr(囊性纤维化跨膜传导调节因子[id1080,ncbi])基因靶标的lamp引物已在其他地方(18)公开。
设计并合成了由荧光标记的fip引物代表的另外的探针,该引物与和猝灭剂连接的互补fd寡核苷酸一起对齐,以确保基于荧光的靶标特异性读出,以在单重/多重测定中检测和区分每种独特的靶标。引物序列提供在表2中。
实例7
靶dna样品
对于qlamp的初步开发,将来自人类sry基因(性别决定区y[id6736,ncbi])和rpa4基因(复制蛋白a4[id29935,ncbi])的200bppcr产物用作靶序列,该产物使用qiaquickpcr纯化试剂盒(凯杰公司)纯化,随后通过sanger进行确认。所使用的引物序列显示在附表2中。首先测试纯化的pcr片段的系列稀释液以优化lamp参数以进行正确定量并表征测试限制。
对人类基因组样品的后续测试利用从成人原代血细胞和来自具有正常雄性(46xy)[gm12877,coriell]和雌性(46xx)[gm12878coriell]核型的b淋巴细胞的良好表征的类淋巴母细胞系分离的dna样品。性染色体比率异常(47xyy、45x0)的样品源自coriell细胞库的相应细胞系(例如,成纤维细胞系,xyy综合征[gm11337])和原始人体组织样品。通过适当的基于柱的制造方案(
为了从基因组dna中释放二级结构,将样品在100℃下孵育范围从30-120sec的不同时间。未处理的完整基因组dna(对照)和预热的dna均用于制备10倍、2倍和1.5倍系列稀释液,并且用lamp对它们进行测试以用于特异性靶标定量。
实例8
lamp反应
使用靶标特异性荧光探针(例如,5’-猝灭剂-fip:fd-荧光团-3’(q-fip:fd-fluo)双链体)进行lamp,这些探针在使用前立即通过混合等体积的200μm5’-猝灭剂-fip(5’-修饰)和200μmfd-荧光团(3’-修饰)以及两倍体积的无核酸酶水以达到每种寡核苷酸的50μm浓度来制备(18)。将混合物在98℃下加热3min以使互补序列对齐,随后缓慢逐渐冷却至室温。
用靶标特异性荧光探针的完全lamp反应含有0.8μmfip和0.4μmq-fip:fd-fluo双链体、1.6μmbip、f3和b3各0.2μm、lf和lb各0.4μm;2.8ubst2.0warmstartdna聚合酶(新英格兰生物实验室(neb));dntp各1mm(neb);dna模板(测试的稀释液);另外补充有6mmmgso4(neb)、1xrox参考染料(0.5μm)的1x等温扩增缓冲液。对于多重反应,将总引物浓度保持为针对标准lamp反应描述的那些,但是其中每组代表总数的1/n,其中n是靶标和相应引物组的数目。
在10μl体积中进行lamp反应。在实验内(测定内)和在独立实验中(测定间)一式两份地或一式三份地分析所有测试的dna样品。将反应板在62℃下孵育60min,并且使用qpcr实时循环仪(steponeplustm实时pcr系统,应用生物系统公司)以30-sec循环步骤以实时模式进行监测。
实例9
数据分析,统计。
使用ct值(反映达到阳性信号阈值的时间)来绘制随参考样品(46xy核型)中每种单独靶标的靶标拷贝数输入负载变化的校准曲线。使用测试样品的每种稀释液的相应扩增ct值来计算那些样品中的输入靶标拷贝数。相对于参考样品稀释液的相应值归一化测试样品的每种稀释液中所获得的靶标拷贝数,以计算靶标拷贝数比率。
通过学生t检验评价同时发生率的均值和方差的显著性,并且确定效应量以及用于一式两份重复和一式三份重复的每个实验内(测定内)以及独立实验之间(测定间)的p值和标准差。
实例10
结果-人类样品中染色体的定性检测
为了确定lamp是否可以定性地检测人类全基因组dna样品中的靶序列,从snp微阵列确认的正常雄性(46xy)和雌性(46xx)的外周血、成纤维细胞系以及羊水(af)样品中分离纯化的基因组dna。使用基于浊度/吸光度和荧光的报告基因测定以及先前采用和描述的检测原理(19)进行lamp,其使用具有不变基因座的对y染色体(sry)和x染色体(rpa4)具特异性的两个独特的单拷贝靶基因的六种探针的引物组。通过实时监测产物扩增期间的荧光增加,使用靶标特异性荧光标签(探针)获得最高质量、灵敏度、特异性和准确性(图1)(18)。在这些条件下,可以仅在15-30min内并且需要少至1-2ng的基因组dna区分具有明显差别的雄性和雌性样品(图1a、图1b、图1d)。
假如原始的或处理的临床af样品含有非常低的dna浓度(在小的测试样品等分试样中数十到几个胎儿基因组拷贝),则在45-60min实现性别鉴别,这取决于样品加工方法(图1c)。
总之,这些初步结果指示,通过基于lamp的反应,可以在对照dna和af样品的存在/不存在分析中快速可靠地定性地测定每种人类dna靶标。尽管这可以作为快速且精确的胚胎和新生儿性别决定的新的有效且方便的工具,如在外生殖器性别不清的情况下,但是有用得多的是可以区分基因组dna样品中不同靶序列的相对拷贝数的定量方法。
实例11
结果-定量靶标检测:分离的靶标。
为了开发一种使用qlamp定量分析靶dna的方法,首先测试了qlamp检测不同浓度的纯化的小dna靶标的能力。从基因组dna模板预扩增对人类x和y性染色体具特异性的两个独特的单拷贝靶序列(分别是rpa4区和sry区)并且用qlamp进行测试。三体性(如21三体(又名唐氏综合症))代表受影响染色体数目增加1.5倍。因此,为了成为有效的筛选或诊断测试,qlamp需要检测靶dna浓度的1.5倍差异。我们制备了范围从1.5x1011至150个拷贝的分离的sry靶标的10倍(图5)、2倍和1.5倍系列稀释液。在整个稀释范围内,10x稀释液的qlamp校准曲线是高度可再现的拷贝(从约1500至约1.5x1011个拷贝)(图2a、图2b;图6)。对于2倍和1.5倍稀释液,分别在1.5x105-1172(最大sd=0.2278)和2.6x104-3424(sd=0.1999)sry靶标拷贝范围内获得了高度可再现的结果(图7)。
为了确定基因组dna的存在是否干扰qlamp,在存在等摩尔基因组拷贝数的缺少sry靶序列的46xx基因组dna的情况下测试系列稀释液。
46xx基因组dna的存在不干扰扩增的准确性和效率,并且仅在扩增过程中引起轻微延迟(图6)。总之,这些数据指示,至少在分离的dna水平上,即使初始输入小至1172靶标拷贝数,也可以容易且可靠地分辨靶标拷贝数的2倍至1.5倍差异。还确认非靶dna本身的存在似乎不影响检测的准确性和精确度。
实例12
结果-定量靶标检测:全基因组样品。
接下来,我们测试了lamp在从外周血样品和细胞培养物中分离的全基因组dna内定量地测量靶标浓度的浓度差异的能力。我们首先评价了基于dna质量和片段化等温扩增(lamp)和定量的靶标可及性如何改变。通过酶消化(非位点特异性核酸酶,如片段酶或微球菌核酸酶)、机械消化(剪切或超声处理以部分消化dna)和dna预热来处理基因组dna样品。用qlamp测试所得样品以优化dna样品加工的其他步骤。显示靶基因组dna的酶消化和机械消化不足并且不能提高qlamp效率、准确性、精确度以正确定量靶序列(图8和图9)。取决于消化的时间和强度,在重复内检测的扩增率和可再现性不足以绘制稀释曲线并检测片段化输入dna的系列稀释液之间的差异(图8),或者至少与原始的(未处理的)dna的扩增率和可再现性相当(图9)。推测起来,这些处理程序引起dna损伤,这损害了qlamp有效扩增靶序列的能力。
我们假设,短暂的热变性步骤可能会从人类基因组dna样品中去除三级dna结构,从而提高lamp的靶标可及性并减少样品之间的差异,从而提高测定的准确性和精确度。我们将基因组dna样品预热至100℃,时间间隔范围从10-180sec,将其扩增率与原始基因组dna和分离的靶标进行比较。如图3所示,预热加热基因组dna样品提高了sry靶标扩增效率、可再现性和准确性。
实例13
结果-性染色体的定量。
我们接下来在源自血细胞和细胞培养物的正常雄性和雌性核型(46xy、46xx)的全基因组dna样品中评估了lamp测量相对x染色体含量的能力。我们的目的是可靠地区分x染色体拷贝数的2倍比率。我们(重复地)制备了每种预热dna样品的范围从85.8ng(26x103个基因组拷贝)至10.7ng(3250个基因组拷贝)的相同的2倍系列稀释液,并且用基于lamp的测定对它们进行测试以定量相对rpa4靶标拷贝数。将对应于达到阳性信号的扩增时间的所获得的ct值用于绘制稀释曲线(图4a)。将46xy样品用作信号归一化的参考样品。根据描述参考46xy的稀释(标准)曲线的等式计算每个稀释点的46xx样品中的rpa4靶标拷贝数(图4a)。在相应的稀释点,相对于46xy样品中的参考拷贝数归一化在46xx样品中检测到的rpa4靶标(即,x染色体)拷贝的计算的数目。计算每个稀释点的一式三份重复的标准差(测定内),并且也相对于参考样品进行归一化。在两次独立实验中重复和再现该测试(测定间);也计算了测定间标准差(图4a、图4b)。因此,lamp可以测量使用从外周血样品中分离的预热基因组dna样品区分46xy和46xx核型的基因组dna样品所需的rpa4的预期1:2相对定量。相比之下,即使在预热步骤之后,源自培养的细胞系的dna仍显示lamp质量差,无法正确靠近和定量rpa4靶标(图10)。每个稀释点的重复信号偏差都特别高,大大降低了分析的准确性。
总的来说,数据证明了lamp定性地检测从外周血、组织和羊水中提取的人类基因组dna样品中的靶序列,更重要的是定量地测量靶序列的相对水平(例如,染色体拷贝数)的有利能力。
实例14
讨论
结果证明,基于qlamp的技术不仅可以成功应用于人类基因组dna样品中的定性靶标检测,还可以成功应用于以高分辨率准确且精确的靶标相对定量。这可以提供染色体拷贝数比率或cnv的正确相对定量。
相比之下,即使在预热步骤之后,源自培养的细胞系的dna仍显示lamp质量差,无法正确靠近和定量rpa4靶标(图10)。每个稀释点的重复信号偏差都特别高,大大降低了分析的准确性。即使当在相对于内部参考(例如,cftr基因)归一化后尝试定量源自不同来源的dna样品(未处理的或以通用方式处理的,例如dna短期预热)(例如,来自血细胞和培养的细胞的46xy和46xxdna对来自培养的细胞的47xyydna对来自冷冻组织的45x0dna)中的相对染色体拷贝数差异时,所获得的结果的变异性/偏差也足够大以用于临床上有意义的用途。原代成熟细胞和培养的分裂和生长细胞经历不同的功能和发育过程,因此基因组dna可以具有特定的确认、配置和修饰。例如,成体原代细胞(例如,血细胞)就恒定发育水平而言都是普适的,而生长的培养细胞群由于去同步化的细胞周期而不均一,因此出现不同的dna状态。我们假设源自不同来源(成熟血细胞、培养的细胞系、结构和内容物不均一的复杂组织)的大的基因组dna可能经历不同的确认改变和修饰,这对于高选择性且具靶标特异性的基于lamp的定量的准确性和精确度可能是至关重要的。尽管如此,我们推想源自特定类型的原代来源(例如类型特异性原代细胞,如血细胞)的dna就确认/修饰状态而言是普适且均一的,并且简单的dna处理(如预热)应该适合于加工dna用于定量lamp。
可以使用qlamp测试用于人类遗传筛查目的的临床样品,例如产前诊断和pgs中的非整倍性和cnv检测。
参考文献
1.lee,h.l.,mcculloh,d.h.,hodes-wertz,b.,adler,a.,mccaffrey,c.andgrifo,j.a.(2015)invitrofertilizationwithpreimplantationgeneticscreeningimprovesimplantationandlivebirthinwomenage40through43.journalofassistedreproductionandgenetics,32,435-444.
2.meberg,a.,hals,j.andthaulow,e.(2007)congenitalheartdefects-chromosomalanomalies,syndromesandextracardiacmalformations.actapaediatr,96,1142-1145.
3.rosenfeld,j.a.,tucker,m.e.,escobar,l.f.,neill,n.j.,torchia,b.s.,mcdaniel,l.d.,schultz,r.a.,chong,k.andchitayat,d.(2015)diagnosticutilityofmicroarraytestinginpregnancyloss.ultrasoundinobstetrics&gynecology:theofficialjournaloftheinternationalsocietyofultrasoundinobstetricsandgynecology,46,478-486.
4.baker,k.,sanchez-de-toledo,j.,munoz,r.,orr,r.,kiray,s.,shiderly,d.,clemens,m.,wearden,p.,morell,v.o.andchrysostomou,c.(2012)criticalcongenitalheartdisease--utilityofroutinescreeningforchromosomalandotherextracardiacmalformations.congenitalheartdisease,7,145-150.
5.charan,p.,woodrow,n.,walker,s.p.,ganesamoorthy,d.,mcgillivray,g.andpalma-dias,r.(2014)high-resolutionmicroarrayintheassessmentoffetalanomaliesdetectedbyultrasound.theaustralian&newzealandjournalofobstetrics&gynaecology,54,46-52.
6.leclercq,s.,lebbar,a.,grange,g.,tsatsaris,v.,letessier,d.anddupont,j.m.(2008)optimizedcriteriaforusingfluorescenceinsituhybridizationintheprenataldiagnosisofcommonaneuploidies.prenataldiagnosis,28,313-318.
7.miller,d.t.,adam,m.p.,aradhya,s.,biesecker,l.g.,brothman,a.r.,carter,n.p.,church,d.m.,crolla,j.a.,eichler,e.e.,epstein,c.j.etal.(2010)
consensusstatement:chromosomalmicroarrayisafirst-tierclinicaldiagnostictestforindividualswithdevelopmentaldisabilitiesorcongenitalanomalies.americanjournalofhumangenetics,86,749-764.
8.vanopstal,d.,boter,m.,dejong,d.,vandenberg,c.,bruggenwirth,h.t.,wildschut,h.i.,deklein,a.andgaljaard,r.j.(2009)rapidaneuploidydetectionwithmultiplexligation-dependentprobeamplification:aprospectivestudyof4000amnioticfluidsamples.europeanjournalofhumangenetics:ejhg,17,112-121.
9.notomi,t.,okayama,h.,masubuchi,h.,yonekawa,t.,watanabe,k.,amino,n.andhase,t.(2000)loop-mediatedisothermalamplificationofdna.nucleicacidsresearch,28,e63.
10.notomi,t.,mori,y.,tomita,n.andkanda,h.(2015)loop-mediatedisothermalamplification(lamp):principle,features,andfutureprospects.journalofmicrobiology,53,1-5.
11.tomita,n.,mori,y.,kanda,h.andnotomi,t.(2008)loop-mediatedisothermalamplification(lamp)ofgenesequencesandsimplevisualdetectionofproducts.natureprotocols,3,877-882.
12.fu,s.,qu,g.,guo,s.,ma,l.,zhang,n.,zhang,s.,gao,s.andshen,z.(2011)applicationsofloop-mediatedisothermaldnaamplification.appliedbiochemistryandbiotechnology,163,845-850.
13.hirayama,h.,kageyama,s.,moriyasu,s.,sawai,k.andminamihashi,a.(2013)embryosexingandsexchromosomalchimerismanalysisbyloop-mediatedisothermalamplificationincattleandwaterbuffaloes.thejournalofreproductionanddevelopment,59,321-326.
14.nogami,h.,tsutsumi,h.,komuro,t.andmukoyama,r.(2008)rapidandsimplesexdeterminationmethodfromdentalpulpbyloop-mediatedisothermalamplification.forensicscienceinternational.genetics,2,349-353.
15.scott,r.t.andtreff,n.r.(2010)us.
16.firth,h.v.,richards,s.m.,bevan,a.p.,clayton,s.,corpas,m.,rajan,d.,vanvooren,s.,moreau,y.,pettett,r.m.andcarter,n.p.(2009)decipher:databaseofchromosomalimbalanceandphenotypeinhumansusingensemblresources.americanjournalofhumangenetics,84,524-533.
17.internationalhapmap,c.,frazer,k.a.,ballinger,d.g.,cox,d.r.,hinds,d.a.,stuve,l.l.,gibbs,r.a.,belmont,j.w.,boudreau,a.,hardenbol,p.etal.(2007)asecondgenerationhumanhaplotypemapofover3.1millionsnps.nature,449,851-861.
18.tanner,n.a.,zhang,y.andevans,t.c.,jr.(2012)simultaneousmultipletargetdetectioninreal-timeloop-mediatedisothermalamplification.biotechniques,53,81-89.
19.zhang,x.,lowe,s.b.andgooding,j.j.(2014)briefreviewofmonitoringmethodsforloop-mediatedisothermalamplification(lamp).biosensors&bioelectronics,61,491-499.
20.nagamine,k.,watanabe,k.,ohtsuka,k.,hase,t.andnotomi,t.(2001)
loop-mediatedisothermalamplificationreactionusinganondenaturedtemplate.clinchem,47,1742-1743.
21.njiru,z.k.(2012)loop-mediatedisothermalamplificationtechnology:towardspointofcarediagnostics.plosnegltropdis,6,e1572.
22.brezina,p.r.andkutteh,w.h.(2015)clinicalapplicationsofpreimplantationgenetictesting.bmj,350,g7611.
23.desanctis,l.andgiuffre,m.(2010)laboratoryinvestigationsingeneticsyndromes:examplesofclinicalapproachintheneonatalunit.minervapediatrica,62,193-195.
24.mcdonald-mcginn,d.m.andzackai,e.h.(2008)geneticcounselingforthe22q11.2deletion.devdisabilresrev,14,69-74.
25.bartlett,l.a.,berg,c.j.,shulman,h.b.,zane,s.b.,green,c.a.,whitehead,s.andatrash,h.k.(2004)riskfactorsforlegalinducedabortion-relatedmortalityintheunitedstates.obstetgynecol,103,729-737.
26.christianson,a.,howson,c.p.andmodell,b.(2006),marchofdimesbirthdefectsfoundation,whiteplains,usa.
27.bardos,j.,hercz,d.,friedenthal,j.,missmer,s.a.andwilliams,z.(2015)anationalsurveyonpublicperceptionsofmiscarriage.obstetgynecol,125,1313-1320.
28.foyouzi,n.,cedars,m.i.andhuddleston,h.g.(2012)cost-effectivenessofcytogeneticevaluationofproductsofconceptioninthepatientwithasecondpregnancyloss.fertilsteril,98,151-155.
29.hirayama,h.,kageyama,s.,takahashi,y.,moriyasu,s.,sawai,k.,onoe,s.,watanabe,k.,kojiya,s.,notomi,t.andminamihashi,a.(2006)rapidsexingofwaterbuffalo(bubalusbubalis)embryosusingloop-mediatedisothermalamplification.theriogenology,66,1249-1256.
30.aoi,y.,hosogai,m.andtsuneda,s.(2006)real-timequantitativelamp(loop-mediatedisothermalamplificationofdna)asasimplemethodformonitoringammonia-oxidizingbacteria.jbiotechnol,125,484-491.
31.cai,t.,lou,g.,yang,j.,xu,d.andmeng,z.(2008)developmentandevaluationofreal-timeloop-mediatedisothermalamplificationforhepatitisbvirusdnaquantification:anewtoolforhbvmanagement.jclinvirol,41,270-276.
32.mori,y.,kitao,m.,tomita,n.andnotomi,t.(2004)real-timeturbidimetryoflampreactionforquantifyingtemplatedna.jbiochembiophysmethods,59,145-157.
- 还没有人留言评论。精彩留言会获得点赞!