蛋白质筛选和检测方法与流程
描述。
背景技术:
蛋白质筛选和蛋白质展示方法是现有技术对表现某些特征(例如,对于靶分子的高亲和力结合)的蛋白质进行鉴定或富集的方法。
在筛选中,逐一分析蛋白质。这是非常费力的,并且限于较低数量的试验。例如,在结合蛋白的筛选中,通过elisa鉴定各结合蛋白候选物,进一步表征阳性elisa命中,例如,通过尺寸排阻色谱、解折叠实验对其进行生物物理学表征,在动物模型中体内检测其治疗潜能。
在展示方法中,经数轮筛选富集整个蛋白质池(源于文库)。对于蛋白质池的处理允许无大量劳动情况下的巨大通量。然而,诸如噬菌体、核糖体或酵母菌展示的展示方法需要表型(蛋白质)和基因型(其编码核酸)之间的物理连锁。因为进行展示(即,噬菌体、核糖体以及编码dna或rna)所需的物理实体大小通常是实际结合分子(例如抗体片段)的超过100倍,这对于大多数分析来说是严重的限制。这不可避免地导致筛选偏差,并将可能的筛选压力限制到小亚组的可想象的筛选压力—当前仅不受展示颗粒巨大尺寸严重影响的筛选压力可应用(例如结合)。
基于上述现有技术,本发明的目的是提供在缺乏物理基因型-表型连锁的情况下从整个蛋白质文库鉴定符合所限定的生物物理学或药理学标准的各蛋白质的方式和方法。此目的通过本说明书的权利要求来实现。
术语和定义
本领域技术人员知道,在本说明书范围内,表示文库大小的数字涉及文库成员的多样性。大于文库ii的文库i对应于包含比文库ii更多数量的独特文库成员的文库i。具有100.000个成员的核酸文库可包含数百万个核酸分子,但是仅100.000个各自由所述文库中独特核酸序列表征的独特文库成员。相似地,具有1.000个成员的多肽文库可包含数百万个多肽分子,但是仅1.000种独特的多肽文库成员。表述“文库的一个成员”涉及可存在于多个相同拷贝的一个特别的文库成员。
在本说明书上下文中,表述“两个核酸序列符合读框”是指第一核酸序列的最后密码子和第二核酸序列的第一密码子之间的碱基对数量可被3除尽。
在本说明书上下文中,表述“多肽与检测标记缔合”、“多肽/检测标记与亲和标记缔合”分别表示前述两个成员均包含在一个一级氨基酸序列、即一个连续的多肽链中。具体地,所述检测标记和所述多肽可由一个或多个氨基酸分离。所述检测标记和所述亲和标记也可由一个或多个氨基酸分离。
在本说明书上下文中,表述“可切割元件”涉及易于被化学试剂或酶方式(例如蛋白酶)切割的肽序列。蛋白酶可为序列特异性性(例如凝血酶)或具有有限的序列特异性(例如胰蛋白酶)。可切割元件i和ii还可包含在检测标记或多肽的氨基酸序列内,特别是当检测标记或多肽的最后氨基酸是k或r的情况。
在本说明书上下文中,术语“亲和标记”涉及与多肽连接的部分,使得能够从生物化学混合物纯化所述多肽。纯化(亲和纯化)基于亲和标记和亲和标记的结合配对物之间的高特异性相互作用(解离常数≤10e-5)。亲和标记可由氨基酸序列组成,或可包含化学部分通过翻译后修饰与其连接的氨基酸序列。通过非限制性实例的方式,亲和标记选自his标记、cbp标记(cbp:钙调素结合蛋白)、cyd标记(cyd:共价但是可解离的norpd肽)、strep标记、strepii标记、flag标记、hpc标记(hpc:蛋白c的重链)、gst标记(gst:谷胱甘肽s转移酶)、avi标记、生物素化标记、myc标记、3xflag标记以及mbp标记(mbp:麦芽糖结合蛋白)。亲和标记另外的实例可见于kimple等,currprotocproteinsci.2013sep24;73:unit9.9。
在本说明书上下文中,术语“深测序”涉及具有≥5x、特别是≥40x的覆盖区(coverage)的数千不同核酸分子的平行测序。术语“覆盖区”涉及在深测序过程中给定的核苷酸被读取的平均次数。
在本说明书上下文中,术语“抗体”以其在细胞生物学和免疫学领域已知的含义使用。全抗体是包含通过二硫键相互连接的至少两个重链(h)和两个轻链(l)的糖蛋白。各重链由重链可变区(h)和重链恒定区(ch)组成。各轻链由轻链可变区(本文缩写为vl)和轻链恒定区(cl)。重链和轻链的可变区包含与抗原相互作用的结合结构域。抗体的恒定区可介导免疫球蛋白与宿主组织或因子的结合,所述宿主组织或因子包括免疫系统的各种细胞(例如效应细胞)和经典补体系统的第一组分。
在本说明书上下文中,术语“纳米抗体”涉及“单结构域抗体”,即由抗体单可变结构域组成的抗体片段。纳米抗体能够选择性结合特异性抗原。其仅具有12–15kda的分子量(harmsen等,appl.microbiol.biotechnol.77(1):13–22)。通常,纳米抗体通过使用所需抗原对单峰骆驼、骆驼、骆马、羊驼或鲨鱼进行免疫和接下来分离编码重链抗体的mrna来获得。纳米抗体还可源自具有四条链的普通鼠或人igg。
在本说明书上下文中,术语“合成抗体(sybody)”涉及合成纳米抗体。合成抗体并非通过使用抗原免疫获得,而是体外选自合成文库。
在本说明书上下文中,术语“富集”涉及增加化合物混合物中某化合物相对量的过程。
在本说明书上下文中,术语“动态编码(flycode)文库”涉及根据本发明所述的氨基酸序列文库,包含多个序列变异体。
在本说明书上下文中,术语“nestlink”涉及其中检测标记与蛋白质文库连接的方法。接下来标记用于对文库中符合所限定的生物物理学或药理学标准的各蛋白质进行鉴定和定量。nestlink结合了筛选和展示程序的关键益处。
在本说明书上下文中,术语“疏水性值”涉及标志肽的预测值。疏水性值根据下述方程式通过krokhin等,molcellproteomics.2004sep;3(9):908-19中描述的方法来计算:
如果h<38,h=kl*(∑rc+0.42r1cnt+0.22r2cnt+0.05r3cnt),
和
如果h≥38,h=kl*(∑rc+0.42r1cnt+0.22r2cnt+0.05r3cnt)–0.3(kl*(∑rc+0.42r1cnt+0.22r2cnt+0.05r3cnt)–38);
如果h<38,h终值=h;
如果h≥38,h终值=h–0.3*(h–38);
其中,h终值是疏水性值,rc是根据下述表格所述的氨基酸类型特有的滞留系数:
氨基酸x的rcnt被定义为:
rxcnt=(∑rc/20)-rxc
n对应于从n末端1开始的检测标记的残基数。kl被定义为:
如果n<10,kl=1-0.027*(10–n)
如果n>20,kl=1-0.014*(n–20)
否则,kl=1。
氨基酸序列从氨基至羧基末端给出。序列位置的大写字母是指单字母密码子中的l氨基酸(stryer,biochemistry,第3版,第21页)。
发明详述
从多肽文库筛选多肽的方法
根据第一方面,提供了从多肽文库筛选多肽的方法,所述方法包括下述步骤:
a.提供第一核酸文库。所述第一核酸文库的各成员包含编码第一多肽文库的成员的多肽编码序列。所述第一核酸文库的各成员不同于所述第一核酸文库的任何其他成员。
b.提供第二核酸文库。所述第二核酸文库包含多个成员。各成员包含编码检测标记的标记编码序列。各检测标记具有下述特征:
i.所述标记通过氨基酸序列表征,所述氨基酸序列不同于由所述第二核酸文库编码的任何其他检测标记的氨基酸序列。
ii.所述标记通过200da和5000da之间的分子量来表征。在某些实施方式中,所述标记通过500da和2500da之间的分子量来表征。在某些实施方式中,所述标记通过900da和2200da之间的分子量来表征。在某些实施方式中,所述标记通过903da和2180da之间的分子量来表征。
iii.所述标记包含第一可切割元件。
ii中给出的质量规格涉及标记被分离后(即,在对第一可切割元件切割之后)的质量。
c.将所述第一核酸文库成员中包含的多肽编码序列插入所述第二核酸文库成员中。由此生成标记核酸文库,所述标记核酸文库编码标记多肽文库。标记多肽文库的各成员包含多肽和检测标记。检测标记与多肽通过第一可切割元件分离。
标记多肽文库是“嵌套文库”,原因是所述第一核酸文库的多肽编码序列“嵌套”在所述第二核酸文库成员中。所述第二核酸文库是所述标记核酸文库的数倍大。所述标记核酸文库是所述第一核酸文库的数倍大。
在标记核酸文库中,第一核酸文库的各多肽编码序列与第二核酸文库的标记编码序列缔合。所述缔合发生在框架内。在标记核酸序列成员引入合适的宿主中之后,多肽编码序列被插入其中将在合适宿主中经受转录和后续翻译的位置。引入细菌细胞可通过转化来实现。引入非细菌细胞可通过转染来实现。本领域技术人员知道,宿主不一定是翻译所需,还可应用体外翻译技术。对于无细胞表达系统的综述,见rosenblum,febslett.2014jan21;588(2):261-8和zemella,chembiochem.2015nov;16(17):2420-31。多肽编码序列和标记编码序列在相同表达序列中转录。
标记核酸文库包含第一核酸文库的所有多肽编码序列,但是仅第二核酸文库的亚组标记编码序列。标记核酸文库的各成员仅包含一个多肽编码序列和一个标记编码序列。仅在标记核酸文库的一个成员中包含各标记编码序列。换言之,标记核酸文库中各标记编码序列是独特的。然而,在标记核酸文库的数个成员中可包含各多肽编码序列(丰余标记)。在某些实施方式中,第一核酸文库的各多肽编码序列与第二核酸文库的至少一个标记编码序列缔合。在某些实施方式中,第一核酸文库的各多肽编码序列与第二核酸文库的至少两个标记编码序列缔合。在某些实施方式中,第一核酸文库的各多肽编码序列与第二核酸文库的至少5个不同标记编码序列缔合。在某些实施方式中,第一核酸文库的各多肽编码序列与第二核酸文库的至少10个不同标记编码序列缔合。在某些实施方式中,第一核酸文库的各多肽编码序列平均与第二核酸文库的10-30个不同标记编码序列缔合。某些实施方式中,第一核酸文库的各多肽编码序列平均与第二核酸文库的约20个不同标记编码序列缔合。
d.从标记核酸文库获得多个核酸序列。具体地,获得用于标记核酸文库各成员的核酸序列。所述多个核酸序列中的各序列包含多肽编码序列和标记编码序列。
基于步骤d中获得的测序信息,生成数据库。所述数据库包含标记多肽文库中所包括的所有多肽序列和所有检测标记。本领域技术人员知道,由于技术原因,所述数据库可不包含标记核酸文库的每一单个成员。所述序列可为核酸序列和/或氨基酸序列的形式。所述数据库包含其中在标记核酸文库中包括第二核酸文库的亚组标记编码序列的信息。所述数据库还可包含其中标记编码序列与给定的多肽编码序列缔合或分别缔合的信息。
e.对于步骤d中获得的标记编码序列编码的各检测标记预测质谱分析裂解模式。本领域技术人员知道,对于分离的检测标记预测裂解模式,即对于已通过切割第一可切割元件游离于其结合多肽的检测标记预测裂解模式。本领域技术人员知道,预测裂解模式还包含预测所分离的检测标记的总质量。
f.标记多肽文库由标记核酸文库表达。由于步骤c中所描述的丰余标记过程,标记多肽文库可包含使用数种不同检测标记而标记的所述第一多肽文库的给定成员(但是每一分子仅一个标记)。优选丰余标记,原因是其有利于通过多重检测标记对第一多肽文库的成员进行明确的检测,和使检测标记对于标记多肽文库的成员的生物物理学特性的潜在影响最小化。另外,丰余是技术原因所需要的:由于一些检测标记可降低表达水平、在样品制备过程中丢失或在反相柱的疏水性窗中未洗脱可检测不到它们,这通过质谱分析来分析。
g.在筛选步骤中筛选标记多肽文库的成员,产生所筛选的多肽。此筛选步骤包括分离符合所限定的生物化学标准的标记多肽文库的那些成员。换言之,将筛选过程应用于标记多肽文库。此筛选过程必须导致蛋白质的物理分离,以致生成和收集物理分离的亚池。根据本发明所述的方法的关键优点是可能的筛选标准的范围远大于蛋白质展示方法中的筛选标准范围。通过非限制性实例的方式,所述标准可选自如下标准:包括以限定的亲和力结合靶分子的能力、多肽在限定条件下的稳定性、某些聚集行为(例如主要作为单体存在)、对蛋白酶的抗性、组织穿透能力、自血流快速或缓慢清除、穿透血脑屏障的能力、以及在肿瘤中聚集的能力。
h.切割第一可切割元件。由此,将检测标记与所筛选的多肽分离,产生分离的检测标记。
i.以下述方式对分离的检测标记进行鉴定和定量:
i.通过质谱分析记录分离的检测标记的裂解模式。裂解模式提供了有关分离的检测标记的片段的质量和疏水性的信息。裂解模式产生有关分离的检测标记的氨基酸序列的信息。
ii.将步骤i中获得的质量和裂解模式与步骤e中预测的质量和裂解模式匹配。由此鉴定分离的检测标记。通过质谱分析获得的信息与通过对标记核酸文库测序获得的信息结合使得对给定的检测标记进行明确鉴定。
可对预测和记录的裂解模式的匹配精确性进行计分,使得对多肽文库成员进行分级。不同筛选条件之间的多肽分级比较可用作多肽不同特性(例如,解离速率、组织分布、构象特异性结合等)的相对测量值。所述比较对于丰余标记多肽文库成员是最精确的,其中计算记录各标记效能的裂解模式差异的平均值。
所预测和记录的裂解模式的匹配精确性的分数可用作筛选后多肽文库成员相对量的测量值。相对量对于丰余标记多肽文库成员是最精确的,其中计算记录各标记的裂解模式的差异的平均值。
j.从步骤d中获得的所述多个核酸序列,筛选包含编码步骤i中所鉴定的所述检测标记的标记编码序列的核酸序列。由此鉴定与步骤i中鉴定的所述检测标记缔合的所述标记多肽文库的成员。
本领域技术人员知道,对于所述标记多肽文库的多个不同成员平行进行步骤g至j。数个多肽的池展示了在步骤g中选择所限定的标志,通过对这些多肽的检测标记的质谱分析鉴定所有这些多肽。本领域技术人员知道,由于技术原因,并非在此步骤中可鉴定每一个单一多肽。
步骤i中进行的质谱分析是定量的,由此,根据本发明所述的方法使得不仅鉴定样品中的多肽而且对样品中所述多肽进行定量。
为了确保丰余和独特的标记,重要的是:
i)第一文库具有有限的、限定的大小。在某些实施方式中,第一核酸文库5至100.000的大小。在某些实施方式中,第一核酸文库具有100至50.000的大小。在某些实施方式中,第一核酸文库具有500至5.000的大小。
ii)在第一文库插入步骤之前第二核酸文库具有103至1011的大小,特别105至1010的大小,更特别106至109的大小,甚至更特别约108的大小。
iii)在插入步骤之后,所选择的多个多肽/标记组合质粒亚组是所述第一核酸文库成员数量的至少3x、特别地至少5x、更特别地至少15x、甚至更特别地至少25x。
iv)所选择的多个多肽/标记组合质粒亚组是所述第二核酸文库的成员数量的小于50%、特别小于5%、更特别小于0.5%,甚至更特别小于0.05%。
文库的大小可通过步骤a之前的多样性限制步骤来控制,其中第一文库作为来自较大前文库的亚组选择。
根据本发明所述的方法使得在缺乏蛋白质展示方法所需的物理基因型-表型连锁的情况下对蛋白质文库进行分析。这消除了具有大物理实体连接于蛋白质文库成员的缺点(例如,噬菌体或核糖体和编码dna或rna)。整个蛋白质文库可作为选择标准的池来筛选,而非如蛋白质筛选通常的情况检测单个蛋白质。然而,尽管处理整个蛋白质池,由于每一单个蛋白质各自被表征,读数与筛选相似。这在结合蛋白开发领域(药物、诊断、研究工具等)特别合适。可一次对数千候选物中一定范围的蛋白质特性进行分析。一个示例问题是:哪些结合蛋白候选物是稳定的、可溶的以及单体的
根据本发明所述的方法使得解决了恰处于蛋白质治疗开发链初始的相关问题:“哪种结合蛋白具有最大的体内治疗潜能
在某些实施方式中,检测标记通过-27和128之间的疏水性值来表征。在某些实施方式中,检测标记通过-1和70之间的疏水性值来表征。在检测标记被分离之后,即,在切割第一可切割元件之后,疏水性值涉及检测标记的质量。疏水性值不包含缔合的亲和标记。
在某些实施方式中,标记多肽文库成员与亲和标记缔合。这样的亲和标记可简化质谱分析之前对所选择的标记多肽文库成员和/或检测标记自身的纯化。在一个一级氨基酸序列中包含亲和标记和标记多肽文库成员。标记多肽文库的各成员包含多肽和检测标记。亲和标记可与多肽或与检测标记缔合。
在某些实施方式中,亲和标记选自his标记、cbp-标记、cyd-标记、strep-标记、strepii-标记、flag-标记、hpc-标记、gst-标记、avi-标记、生物素化标记、myc-标记、3xflag标记以及mbp-标记。
在某些实施方式中,检测标记与亲和标记缔合。在这些情况下,亲和标记位于检测标记的c末端。此配置具有下述优点,即,检测标记被保护免于肽酶降解,确保在蛋白质纯化过程中仅与完整检测标记缔合的非降解多肽倍分离。本领域技术人员知道,表述“亲和标记位于检测标记的c末端”不一定意味着亲和标记位于检测标记的紧靠c末端,但是可具有分离亲和标记和检测标记的数个氨基酸的连接子。
在某些实施方式中,亲和标记与所述检测标记通过第二可切割元件分离,所述第二可切割元件在步骤i前被切割。由此,仅不具有缔合的亲和标记的检测标记通过质谱分析进行分析。
检测标记的质量和裂解模式说明涉及在其与缔合的多肽和亲和标记分离之后(即在第一和第二可切割元件的切割之后)的质量和裂解模式。本领域技术人员知道,在其中质谱分析之前检测标记未与缔合的亲和标记分离的情况下,这会影响质谱分析的结果。如果所有检测标记与相同的亲和标记缔合,质量和裂解模式的变化可被解释,由此可鉴定检测标记,但是不如其中检测标记通过第二可切割元件与亲和标记分离的情况那么有效明确。
在某些实施方式中,亲和标记是his标记。
在某些实施方式中,步骤h包括通过与电喷雾电离质谱偶联的液相色谱(lc-mc)分析分离的检测标记。在某些实施方式中,此步骤包括反相液相色谱。通过反相色谱根据检测标记的疏水性分开所分离的检测标记,以减小样品复杂性。接下来,通过质谱分析记录其质量和肽裂解模式。
在某些实施方式中,步骤d包括以≥5x的覆盖区对完整的标记表达文库进行测序。在某些实施方式中,步骤d包含对标记表达文库进行深测序。
在某些实施方式中,步骤d包括将多肽编码序列和标记核酸文库中包含的标记编码序列一起插入测序载体。深测序通常包括pcr扩增步骤。本发明的发明人注意到,pcr扩增导致标记文库成员的基因片段之间显著数量的重组事件。由此,他们构建了一组深测序质粒,所述深测序质粒允许通过限制酶消化和接合进行深测序所需的序列元件的连接,由此消除深测序之前对嵌套文库pcr扩增的需要。
在某些实施方式中,分离的检测标记由5至30个连续的氨基酸组成,包含一个或仅一个具有正电荷侧链的氨基酸。在某些实施方式中,分离的检测标记由7至21个连续的氨基酸组成,包含一个或仅一个具有正电荷侧链的氨基酸。在某些实施方式中,分离的检测标记由11至15个连续的氨基酸组成,包含一个或仅一个具有正电荷侧链的氨基酸。
在某些实施方式中,具有正电荷侧链的氨基酸位于分离的检测标记的c末端。在某些实施方式中,具有正电荷侧链的氨基酸选自精氨酸(r)和赖氨酸(k)。在某些实施方式中,具有正电荷侧链的氨基酸是位于分离的检测标记c末端的精氨酸(r)。
本领域技术人员知道,除了具有正电荷侧链的氨基酸以外,中性ph下分离的检测标记携带另一正电荷,其是分离的检测标记n末端的伯胺。
在某些实施方式中,分离的检测标记包含选自序列元件i的集合的序列元件i,其中所述序列元件i由5至10个氨基酸、特别7个氨基酸组成,各氨基酸彼此独立地选自a、s、t、n、q、d、e、v、l、i、f、y、w、g以及p。
在某些实施方式中,具有正电荷侧链的一个和仅一个氨基酸位于分离的检测标记的c末端,其余氨基酸独立地选自a、s、t、n、q、d、e、v、l、i、f、y、w、g以及p。在某些实施方式中,具有正电荷侧链的一个和仅一个氨基酸是位于分离的检测标记的c末端的r。
分离的检测标记可最佳由质谱检测,特别是lc-ms(与esi-ms偶联的反相液相色谱)。在检测标记的设计中省略氨基酸c和m,原因是它们易于氧化。在序列元件i中省略氨基酸k、r以及h,原因是它们会将另外的具有正电荷侧链的氨基酸添加至标记上,这是不想要的,因为在esi-ms过程中标记携带另外的电荷,这落在最佳检测范围之外。k和r会添加另外的酪氨酸切割位点至标记序列中,这是不想要的。
将k添加至检测标记的氨基酸序列会增加另一伯胺,这会使得使用nhs化学方法通过质谱分析相对和绝对定量对检测标记通过标记进行标志复杂。
在某些实施方式中,分离的检测标记包含:
a.序列元件i,其中序列元件i由5至10个、特别7个连续的氨基酸组成,各氨基酸彼此独立地选自a、s、t、n、q、d、e、v、l、i、f、y、w、g以及p;和
b.序列元件ii,所述序列元件ii选自seqidno01(wr)、seqidno02(wlr)、seqidno03(wqsr)、seqidno04(wltvr)以及seqidno05(wqeggr)。
在某些实施方式中,分离的检测标记由下述组成:
a.序列元件iii:gs;
b.序列元件i,其中序列元件i由5至10个、特别7个连续的氨基酸组成,各氨基酸彼此独立地选自a、s、t、n、q、d、e、v、l、i、f、y、w、g以及p;和
c.序列元件ii,所述序列元件ii选自seqidno01(wr)、seqidno02(wlr)、seqidno03(wqsr)、seqidno04(wltvr)以及seqidno05(wqeggr)。
序列元件从n末端至c末端的次序是:序列元件iii、序列元件i、序列元件ii。这些检测标记落在903da和2180da之间的治疗范围内,这对于通过esi-ms的灵敏检测是最佳的。在生理ph及其以下的情况,分离的检测标记携带两个正电荷,即,在c末端的r和n末端伯胺。位于分离的检测标记c末端的正电荷有利于用于质谱检测的标记的电离,用作独特的酪氨酸裂解位点。具有c末端精氨酸或赖氨酸的肽特别可通过质谱分析良好检测(有利的电离特性)。在各分离的检测标记中,n末端胺仅为伯胺,其用于通过nhs化学方法进行胺偶联。这允许连接标记用于定量质谱仪进行例如itraq(用于相对和绝对定量的同量异序标记)。对检测标记进行工程化以展示一定范围的疏水性,所述一定范围的疏水性理想地适于通过标准反相色谱柱进行肽分离。
在某些实施方式中,第一核酸文库中包含的所有序列元件i一起组成序列元件i的集合。在序列元件i的集合中,各氨基酸以表1中指明的频率存在。
表1
在某些实施方式中,所述第一和/或所述第二可切割元件是或包含蛋白酶识别序列。在某些实施方式中,所述第一和所述第二可切割元件均是或包含蛋白酶识别序列。
在某些实施方式中,所述第一可切割元件是或包含凝血酶识别序列,和/或所述第二可切割元件是或包含胰蛋白酶识别序列。
多肽集合
根据第二方面,提供多肽集合。所述多肽集合的各成员与检测标记缔合。在某些实施方式中,述多肽集合的各成员与至少一种检测标记缔合。表述“与至少一种检测标记缔合”是指多肽集合的各成员可与超过一种的检测标记缔合,但是每一多肽分子仅一种标记。换言之,多肽集合可包含与检测标记x缔合的多肽a和与检测标记y缔合的多肽a,而非与检测标记x和y都缔合的多肽a。在某些实施方式中,多肽集合的各成员与至少两种检测标记缔合。在某些实施方式中,多肽集合的各成员与至少五种检测标记缔合。在某些实施方式中,多肽集合的各成员与至少十种检测标记缔合。在某些实施方式中,多肽集合的各成员与至少二十种检测标记缔合。各检测标记具有下述特性:
a.所述标记通过氨基酸标记序列来表征,所述氨基酸标记序列不同于由多个表达载体编码的任何其他检测标记的氨基酸序列。
b.所述标记通过200da和5000da之间的分子量来表征。在某些实施方式中,所述标记通过500da和2500da之间的分子量来表征。在某些实施方式中,所述标记通过900da和2200da之间的分子量来表征。在某些实施方式中,所述标记通过903da和2180da之间的分子量来表征。
c.所述标记与所述多肽集合的所述成员通过第一可切割元件分离。
在本发明第二方面的某些实施方式中,检测标记通过-27和128之间的疏水性值来表征。在某些实施方式中,检测标记通过-1和70之间的疏水性值来表征。
在本发明第二方面的某些实施方式中,多肽集合的成员与亲和标记缔合。
在本发明第二方面的某些实施方式中,检测标记与亲和标记缔合。亲和标记和检测标记包含在相同的一级氨基酸序列中。亲和标记与检测标记通过第二可切割元件分离。检测标记可通过第二可切割元件的切割而与亲和标记分离。在某些实施方式中,亲和标记选自his标记、cbp-标记、cyd-标记、strep-标记、strepii-标记、flag-标记、hpc-标记、gst-标记、avi-标记、生物素化标记、myc-标记、3xflag标记以及mbp-标记。在某些实施方式中,亲和标记是his标记。
在本发明第二方面的某些实施方式中,分离的检测标记由5至30个连续的氨基酸组成,并包含一个和仅一个具有正电荷的侧链。在某些实施方式中,分离的检测标记由7至21个连续的氨基酸组成,并包含一个和仅一个具有正电荷的侧链。在某些实施方式中,分离的检测标记由11至15个连续的氨基酸组成,并包含一个和仅一个具有正电荷的侧链。在某些实施方式中,具有正电荷侧链的氨基酸位于分离的检测标记的c末端。在某些实施方式中,具有正电荷侧链的氨基酸选自精氨酸(r)和赖氨酸(k)。在某些实施方式中,具有正电荷侧链的氨基酸是位于分离的检测标记的c末端的精氨酸(r)。
在本发明第二方面的某些实施方式中,检测标记包含:
a.序列元件i,其中序列元件i由5至10个、特别7个连续的氨基酸组成,各氨基酸彼此独立地选自a、s、t、n、q、d、e、v、l、i、f、y、w、g以及p;和
b.序列元件ii,所述序列元件ii选自seqidno01(wr)、seqidno02(wlr)、seqidno03(wqsr)、seqidno04(wltvr)以及seqidno05(wqeggr)。
检测标记
根据第三方面,提供肽检测标记,所述肽检测标记被设计用于通过质谱分析的最佳检测。检测标记由4至20个氨基酸组成,具有下述特征:
a.检测标记仅包含一个具有正电荷侧链的氨基酸;
b.检测标记通过200da和5000da之间的分子量来表征。在某些实施方式中,检测标记通过500da和2500da之间的分子量来表征。在某些实施方式中,检测标记通过900da和2200da之间的分子量来表征。在某些实施方式中,标记通过903da和2180da之间的分子量来表征。
在本发明第三方面的某些实施方式中,检测标记由7至18个氨基酸组成,在本发明第三方面的某些实施方式中,检测标记由11至15个氨基酸组成。
在本发明第三方面的某些实施方式中,检测标记基本由下述组成:
a.序列元件i,其中序列元件i由5至10个、特别7个连续的氨基酸组成,各氨基酸彼此独立地选自a、s、t、n、q、d、e、v、l、i、f、y、w、g以及p;和
b.序列元件ii,所述序列元件ii选自seqidno01(wr)、seqidno02(wlr)、seqidno03(wqsr)、seqidno04(wltvr)以及seqidno05(wqeggr)。
检测标记集合
根据另一方面,提供肽标记的集合。根据本发明第三方面,肽标记集合包含肽标记。肽标记集合中包含的各检测标记由4至20个氨基酸组成,通过不同于所述检测标记集合中包含的任何其他检测标记的氨基酸序列的氨基酸序列来表征。在某些实施方式中,各检测标记由7至18个氨基酸组成。在某些实施方式中,各检测标记由11至15个氨基酸组成。在某些实施方式中,肽标记集合包含至少96种肽标记。在某些实施方式中,肽标记集合包含至少500.000种肽标记。在某些实施方式中,肽标记集合包含至少107种的肽标记。在某些实施方式中,肽标记集合包含约108种肽标记。
在本发明此方面的某些实施方式中,检测标记包含仅一个具有正电荷侧链的氨基酸,其余的氨基酸选自a、s、t、n、q、d、e、v、l、i、f、y、w、g以及p。
在本发明此方面的某些实施方式中,所述标记通过-27和128之间的疏水性值来表征。在某些实施方式中,检测标记通过-1和70之间的疏水性值来表征。
在本发明此方面的某些实施方式中,检测标记与亲和标记缔合。在某些实施方式中,亲和标记选自his标记、cbp-标记、cyd-标记、strep-标记、strepii-标记、flag-标记、hpc-标记、gst-标记、avi-标记、生物素化标记、myc-标记、3xflag标记以及mbp-标记。在某些实施方式中,亲和标记是his标记。亲和标记和检测标记包含在相同一级氨基酸序列中。亲和标记通过可切割元件与检测标记分离。
在本发明此方面的某些实施方式中,检测标记基本由下述组成:
a.序列元件i,其中所述序列元件i由5至10个、特别7个连续的氨基酸组成,各氨基酸彼此独立地选自a、s、t、n、q、d、e、v、l、i、f、y、w、g以及p;和
b.序列元件ii,所述序列元件ii选自seqidno01(wr)、seqidno02(wlr)、seqidno03(wqsr)、seqidno04(wltvr)以及seqidno05(wqeggr)。
质粒载体集合
根据又一方面,提供了质粒载体集合。所述质粒载体集合的成员包含编码检测标记的核酸序列。各检测标记由4至20个氨基酸组成,通过不同于由所述质粒载体集合编码的任何其他检测标记的氨基酸序列的氨基酸序列来表征。在某些实施方式中,各检测标记由7至18个氨基酸组成。在某些实施方式中,各检测标记由11至15个氨基酸组成。在某些实施方式中,质粒载体集合包含至少96种载体。在某些实施方式中,质粒载体集合包含至少500.000种质粒载体。在某些实施方式中,质粒载体集合包含至少107种质粒载体。在某些实施方式中,质粒载体集合包含约108种质粒载体。
在本发明此方面的某些实施方式中,检测标记仅包含一个具有正电荷侧链的氨基酸。
在本发明此方面的某些实施方式中,检测标记通过200da和5000da之间的分子量来表征。在某些实施方式中,检测标记通过500da和2500da之间的分子量来表征。在某些实施方式中,检测标记通过900da和2200da之间的分子量来表征。在某些实施方式中,检测标记通过903da和2180da之间的分子量来表征。
在本发明此方面的某些实施方式中,所述标记通过通过-27和128之间的疏水性值来表征。在某些实施方式中,检测标记通过-1和70之间的疏水性值来表征。
在本发明此方面的某些实施方式中,检测标记与亲和标记缔合。在某些实施方式中,亲和标记选自his标记、cbp-标记、cyd-标记、strep-标记、strepii-标记、flag-标记、hpc-标记、gst-标记、avi-标记、生物素化标记、myc-标记、3xflag标记以及mbp-标记。在某些实施方式中,亲和标记是his标记。亲和标记和检测标记包含在相同一级氨基酸序列中。亲和标记通过可切割元件与检测标记分离。
在本发明此方面的某些实施方式中,检测标记基本由下述组成:
a.序列元件i,其中所述序列元件i由5至10个、特别7个连续的氨基酸组成,各氨基酸彼此独立地选自a、s、t、n、q、d、e、v、l、i、f、y、w、g以及p;和
b.序列元件ii,所述序列元件ii选自seqidno01(wr)、seqidno02(wlr)、seqidno03(wqsr)、seqidno04(wltvr)以及seqidno05(wqeggr)。
在本发明此方面的某些实施方式中,质粒载体集合的各成员包含:
a.负筛选盒,所述负筛选盒5'侧邻第一限制性核酸内切酶位点,3'侧邻第二限制性核酸内切酶位点;
b.位于所述第一限制性核酸内切酶位点5'的启动子;
c.位于所述第二限制性核酸内切酶位点3'的编码检测标记的核酸标记序列。在某些实施方式中,编码检测标记的核酸序列和所述第二限制性核酸内切酶位点由少于100个碱基对分离。在某些实施方式中,编码检测标记的核酸序列和所述第二限制性核酸内切酶位点由少于50个碱基对分离。在某些实施方式中,编码检测标记的核酸序列和所述第二限制性核酸内切酶位点由约20个碱基对分离。在某些实施方式中,位于编码检测标记的核酸序列和所述第二限制性核酸内切酶位点之间的碱基对编码第一可切割元件。
在本发明此方面的某些实施方式中,质粒载体集合的各成员包含:
a.编码检测标记的核酸标记序列,所述核酸标记序列在相同阅读框中与编码多肽的核酸序列缔合;
b.多样性元件,所述多样性元件包含不全同的碱基以防止测序过程中信号超负荷;
c.用于测序引物结合的引物结合位点;
d.索引元件,所述索引元件包含用于多路技术的数个限定的核酸序列中的一个序列;
e.接头元件,所述接头元件在测序过程中固定dna分子;
f.侧邻元件a-e的两个限制性核酸内切酶位点,所述限制性核酸内切酶位点在测序之前从质粒载体释放dna片段。
先前实施方式中描述的质粒载体用作深测序质粒。优选地,这些载体不包含亲和标记以减少待测序片段的长度。
蛋白质检测方法
根据另一方面,提供一种蛋白质检测方法,所述方法包括下述步骤:
a.提供编码多肽文库的核酸文库。多肽文库中包含的各多肽与检测标记缔合。多肽和检测标记包含在相同的一级氨基酸序列中。各检测标记具有下述特性:
i.所述标记通过氨基酸序列来表征,所述氨基酸序列不同于由核酸文库编码的任何其他检测标记的氨基酸序列。
ii.所述标记通过200da和5000da之间的分子量来表征。在某些实施方式中,所述标记通过500da和2500da之间的分子量来表征。在某些实施方式中,所述标记通过900da和2200da之间的分子量来表征。在某些实施方式中,所述标记通过903da和2180da之间的分子量来表征。
iii.所述标记与缔合的多肽通过第一可切割元件分离。
核酸文库编码的各检测标记就核酸文库编码的任何其他检测标记而言是独特的。多肽文库中包含的各多肽与至少一种检测标记缔合。在某些实施方式中,多肽文库中包含的各多肽与至少两种检测标记缔合。在某些实施方式中,多肽文库中包含的各多肽与至少五种检测标记缔合。在某些实施方式中,多肽文库中包含的各多肽与至少十种检测标记缔合。在某些实施方式中,多肽文库中包含的各多肽与约二十种检测标记缔合。各多肽分子仅包含一种检测标记。
b.提供数据库。所述数据库包含下述信息:
i.多个核酸和/或氨基酸序列。多个序列包含核酸文库所有成员的序列。各序列包含指明多肽的序列和指明检测标记的序列。
ii.用于核酸文库编码的各检测标记的质谱分析裂解模式。
c.从核酸文库表达多肽文库。
d.在筛选步骤中筛选多肽文库成员,产生所筛选的多肽。
e.切割第一可切割元件。由此检测标记与所筛选的多肽分离,产生分离的检测标记。
f.以下述方式鉴定分离的检测标记:
i.通过质谱分析记录分离的检测标记的裂解模式。
ii.将步骤i中获得的裂解模式与在提供的数据库中预测的裂解模式匹配。由此鉴定分离的检测标记。通过质谱分析获得的信息与通过对标记核酸文库测序获得的信息组合使得对给定的检测标记进行明确鉴定。
g.从所述数据库中包含的所述多个序列筛选步骤f中鉴定的指明所述检测标记的序列。由此鉴定与步骤f中鉴定的所述检测标记缔合的所述多肽文库的成员。
在某些实施方式中,所述多肽文库的各成员与亲和标记缔合。在某些实施方式中,各检测标记与亲和标记缔合。
在某些实施方式中,亲和标记选自his标记、cbp-标记、cyd-标记、strep-标记、strepii-标记、flag-标记、hpc-标记、gst-标记、avi-标记、生物素化标记、myc-标记、3xflag标记和mbp-标记。
在某些实施方式中,亲和标记与所述检测标记通过第二可切割元件分离,所述第二可切割元件在步骤f之前被切割。由此,通过质谱分析仅分析无缔合的亲和标记的检测标记。
检测标记的质量和裂解模式说明涉及在检测标记与缔合的多肽和亲和标记分离之后(即在第一和第二可切割元件)的质量和裂解模式。本领域技术人员知道,在其中质谱之前检测标记未与缔合的亲和标记分离的情况下,这会影响质谱分析的结果。由于所有检测标记与相同的亲和标记缔合,可解释质量和裂解模式的改变,由此仍可能鉴定检测标记,但是不如其中检测标记通过切割第二可切割元件与亲和标记分离的情况那么有效明确。
在某些实施方式中,亲和标记是his标记。
本领域技术人员知道,对多肽文库的多个不同成员平行进行步骤d至g。在步骤g中选择具有数个多肽的池,所有这些多肽通过对其检测标记的质谱分析来鉴定。本领域技术人员知道,由于技术原因,并非每一单个多肽均可在此步骤中鉴定。
步骤f中进行的质谱分析是定量的,由此,根据本发明所述的方法使得不仅鉴定样品中的多肽而且还对此多肽进行定量。
使多肽与独特的检测标记缔合的方法
根据又一方面,提供了一种使多肽与独特的检测标记缔合的方法,所述方法包括下述步骤:
a.提供第一核酸文库,第一核酸文库的各成员包含编码第一多肽文库成员的多肽编码序列。
b.提供第二核酸文库,第二核酸文库的各成员包含编码检测标记的标记编码序列。各检测标记具有下述特性:
i.所述标记通过氨基酸序列表征,所述氨基酸序列不同于第二核酸文库编码的任何其他检测标记的氨基酸;
ii.所述标记通过200da和5000da之间的分子量表征。在某些实施方式中,所述标记通过500da和2500da之间的分子量来表征。在某些实施方式中,所述标记通过900da和2200da之间的分子量来表征。在某些实施方式中,所述标记通过903da和2180da之间的分子量来表征。
c.将第一核酸文库成员中包含的多肽编码序列插入第二核酸文库成员中。由此生成多个多肽—标记组合质粒。
第一核酸文库具有5至100.000的大小。在某些实施方式中,第一核酸文库具有100至50.000的大小。在某些实施方式中,第一核酸文库具有500至5.000的大小。
第二核酸文库具有103至1011的大小。在某些实施方式中,第二核酸文库具有105至1010的大小。在某些实施方式中,第二核酸文库具有106至109的大小。在某些实施方式中,第二核酸文库具有约108的大小。
在多个多肽/标记组合质粒中,第一核酸文库的各多肽编码序列与第二核酸文库的标记编码序列缔合。所述缔合发生在相同阅读框中。
d.筛选多个多肽—标记组合质粒的亚组。此筛选步骤包括筛选限定量的克隆,其中各克隆包含多个多肽—标记组合质粒的一个成员。由此生成编码标记多肽文库的标记核酸文库。标记多肽文库的各成员包含多肽和检测标记。各标记仅包含在标记多肽文库的一个成员中。换言之,标记多肽文库中的各检测标记是独特的。然而,各多肽可包含在标记多肽文库的数个成员中(丰余标记)。
在某些实施方式中,各多肽与至少一种检测标记缔合。在某些实施方式中,各多肽与至少两种检测标记缔合。在某些实施方式中,各多肽与至少五种检测标记缔合。在某些实施方式中,各多肽与至少十种检测标记缔合。在某些实施方式中,各多肽与约二十种检测标记缔合。
在本发明此方面的某些实施方式中,多个多肽—标记组合质粒的所筛选亚组是第一核酸文库成员数量的至少10x。在某些实施方式中,多个多肽—标记组合质粒的所筛选亚组是第一核酸文库成员数量的至少20x。
在本发明此方面的某些实施方式中,多个多肽—标记组合质粒的所筛选亚组是第二核酸文库成员数量的少于50%。在本发明此方面的某些实施方式中,多个多肽—标记组合质粒的所筛选亚组是第二核酸文库成员数量的少于5%。在本发明此方面的某些实施方式中,多个多肽—标记组合质粒的所筛选亚组是第二核酸文库成员数量的少于0.05%。
通过选择多个多肽—标记组合质粒的亚组的最佳大小,确保标记多肽文库中,各检测标记是独特的(仅存在一次),但是各多肽存在数次,每次与不同检测标记缔合。
虽然用于单个可分离特征的替代方案在本文称之为"实施方式",应当理解这些替代方案可被自由地组合以形成本文公开的本发明的分立实施方式。
通过下述实施例和附图进一步说明本发明,从这些实施例和附图可以得到进一步的实施方式和优点。这些实施例旨在说明本发明,但不限制其范围。
附图说明
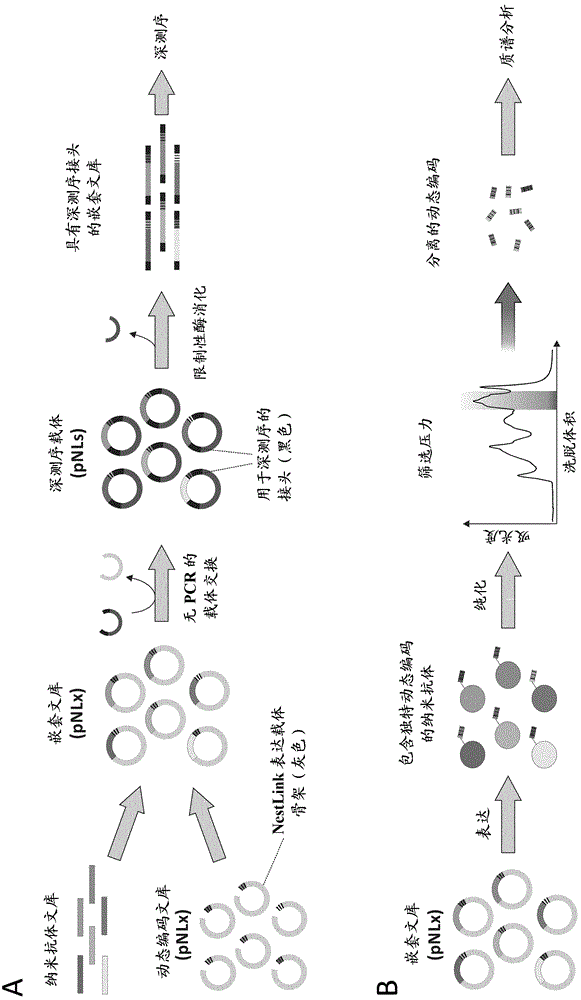
图1显示nextlink技术的概况。a)在表达载体pnlx上编码的动态编码文库内嵌套有纳米抗体文库。接下来,通过限制性酶消化解离动态编码纳米抗体序列,并插入pnl中,其导致所需接头序列连接用于深测序。然后,通过限制性酶消化解离与动态编码纳米抗体连接的接头,并以线性形式使其经受深测序。b)表达并纯化pnlx中编码的嵌套文库。应用筛选压力(在此特殊情况下,通过尺寸排阻色谱筛选具有纳米抗体单体表观分子量的蛋白质),并通过蛋白酶裂解分离所筛选的纳米抗体的动态编码。c)深测序数据使得生成将所有动态编码分配至其相应纳米抗体的数据库。拼接各纳米抗体的动态编码。先前分离的动态编码(见b)经受lc-ms,并生成记录的ms/ms数据的峰系列。针对包含拼接的动态编码的数据库检索ms/ms数据,这允许对筛选的纳米抗体进行鉴定和相对定量。
图2显示在文库插入之前(上游字符串)和之后(下游字符串)用于nestlink技术的相关质粒设计。a)用于针对靶分子的纳米抗体噬菌体展示筛选的噬菌粒。所述噬菌粒携带两个sapi限制性酶切位点,其允许纳米抗体文库插入,和在通过噬菌体展示富集后其有效转移至nestlink表达载体pnlx。b)携带约108个变异体的动态编码多样性的nestlink表达载体pnlx。sapi位点被设计成当纳米抗体文库插入时消失。动态编码纳米抗体可特别通过sfii限制性酶切位点从表达载体解离。sfi位点的位置确保与相应动态编码连接的完整纳米抗体的深测序,单通过排除丰余序列(例如pelb和his标记)使深测序阅读长度最小化。c)生成具有不同索引的一组深测序载体(pln),各所述深测序载体携带用于illuminamiseq测序的所有必要序列。通过sfi限制性酶切和接合将动态编码纳米抗体插入此载体。接下来,通过bseri限制性酶切它们作为包含所有miseq接头区的线性片段被释放。以此方式,不需要pcr来产生用于miseq分析的dna片段,这会导致纳米抗体—动态编码序列中的重组事件,由此破坏动态编码和纳米抗体序列之间的的连接。d)深测序接头还可经由与pnlx中编码的sfii限制性酶切位点互补的合适单链悬突通过合成的双链接头寡核苷酸来连接。
图3显示使用lc-ms通过动态编码对ploi成员进行绝对定量。动态编码合成抗体的七个已知定量(x轴)被掺加至分别包含来自大肠杆菌(e.coli)或耻垢分枝杆菌(m.smegmatis)的裂解物的两种不同样品(背景)。如深测序所测定,动态编码合成抗体在0.2、0.4、1.3、4.1、8.5、18.0以及27.5吸光单位(280nm)下被掺加,包含28、56、112、56、112、84以及112个动态编码。通过lc-ms分析分离的动态编码。使用progenesis软件总计来自各合成抗体的所有动态编码的ms1强度。
图4显示通过nestlink鉴定来自1’080个候选结合蛋白的表现最佳解离速率的合成抗体。a:将溶液(sec)中与生物素化靶蛋白共洗脱的单体合成抗体固定于两个当量的链霉亲和素琼脂糖凝胶柱。使用缓冲液洗涤一个柱子,另一个柱子通过过量的非生物素化靶洗涤3分钟。接下来,分离其余结合的合成抗体的动态编码,通过lc-ms1强度定量。b:对于各池成员测定lc-ms1强高度(所有动态编码的总计),对于各合成抗体(x轴)在y轴上绘出两个柱子之间比率。未表达的、非单体的或未结合溶液中靶的合成抗体在任一柱子上均不可检测到,原因是它们作为原理证明试验中描述的预筛选过程的结果被去除(合成抗体320–1’080)。弱结合合成抗体仅在缓冲液洗涤之后可检测到,但在与过量靶竞争的情况下不可检测到(合成抗体187–320)。在两个柱子上均检测到合成抗体1-186,根据其解离速率分类。用于下游应用的最有前景的合成抗体是具有最慢解离速率导致比率接近1的那些。c:nestlink读数和各挑选的合成抗体的spr试验之间的相关性。合成b)中分析的11种合成抗体的dna序列(基因合成),表达、纯化相应的结合蛋白,通过表面等离振子共振逐个分析。spr数据在洗涤3分钟后在x轴作为残余结合信号(作为解离速率的测量值)和在y轴作为b)所示的通过nestlink测定的合成抗体比率绘出。
图5:来自免疫的羊驼的3’469种纳米抗体的分析及其鉴定,其在溶液中表现最强的抗原结合。在消除具有不良表达水平(步骤1)和可溶性(步骤2,单体抗体的筛选)的那些池成员之后,在三个不同化学计量下将池的单体级分与膜蛋白抗原一起温育,并通过sec分析。步骤1之后(报告各池成员表达水平)、步骤2(报告各池成员的可溶性)以及在步骤3从所有靶/复合物峰收集lc-ms样品。如通过总计各纳米抗体的所有ms1强度来确定的,圆图表现在筛选过程的不同阶段池中各纳米抗体的相对量(总体淡灰色表示的非结合蛋白或弱结合蛋白,池成员的总量对应于100%)(100%=所有纳米抗体的所有动态编码的所有ms1强度总和)。如步骤3所预期的,池抗原比率增加导致许多池结合成员对有限数量抗原的内部竞争性增加。由此,具有最强亲和性的池成员的级分以对有限表位较高的竞争性增加。
图6:针对纯化的外膜蛋白靶,来自通过体外筛选(步骤1)生成的池的1’456种合成抗体的分析,用于感兴趣的革兰氏阴性细菌的细胞表面结合(步骤2)。在步骤2(nestlink)中,首先将那些具有不良表达水平和可溶性的池成员从总体中消除,之后使用4种不同感兴趣的菌株进行4个不同的拉下试验。在通过洗涤去除未与细胞以高亲和力结合的池成员之后,通过lc-ms分离和分析池的所有动态编码。然后,各合成抗体的所有动态编码的所有ms1强度的总和可用作各靶细胞的池中各合成抗体的相对浓度的测量值。这允许对于各合成抗体(x轴)在4种细胞类型的每种中报告其相对浓度(相对于整个池)的明确的细胞特异性读数(b)。出于清楚的原因,b中仅显示所有分析的合成抗体的25%。
实施例
动态编码序列文库
具有短dna编码的肽的随机文库被设计成通过质谱分析(ms)、特别是通过lc-ms(与esi-ms偶联的反相液相色谱)可最佳检测到。所述肽落在903da和2180da之间的质量范围内,这对于通过esi-ms的灵敏检测是最佳的。在生理ph及其以下的情况,动态编码携带两个正电荷,即,在c末端的r和n末端伯胺。位于动态编码c末端的正电荷有利于用于质谱检测的标记的电离,用作独特的酪氨酸裂解位点。具有c末端精氨酸或赖氨酸的肽特别可通过质谱分析良好检测(有利的电离特性)。在各动态编码中,n末端胺仅为伯胺,其用于通过nhs化学方法进行胺偶联。这允许连接标记用于定量质谱仪进行例如itraq(用于相对和绝对定量的同量异序标记)。对动态编码进行工程化以展示一定范围的疏水性,所述一定范围的疏水性理想地适于通过标准反相色谱柱进行肽分离。
动态编码文库由两部分加侧邻氨基酸组成,所述侧邻氨基酸是恒定的,即,n末端的gs和c末端的r。n末端“gs”序列是凝血蛋白酶裂解位点的部分,在裂解之后仍处于动态编码。
部分1:条形码区包含7个连续的随机氨基酸位置。氨基酸的平均频率在上述表1中给出(%)。
并非所有二十种天然氨基酸均存在于条形码(c、m、k、r、h以及i丢失)中。c和m被省略,原因是它们易于氧化。k、r以及h被省略,原因是它们会将另外的正电荷添加至动态编码序列上,这是不想要的,因为在esi-ms检测过程中肽携带另外的电荷,这落在最佳检测范围之外。k和r会添加另外的酪氨酸切割位点至动态编码中,这是不想要的。k会添加另一伯胺,这会使得通过化学方法进行肽标记复杂。异亮氨酸被省略,原因是它不能通过质量与亮氨酸区分。
部分2:以5种不同变异体构建c末端,所述变异体在动态编码文库中具有相同频率,并且均以r结束。它们也都无c、m、k、h以及i。由此,动态编码由最少11个氨基酸组成,由最多15个氨基酸组成(gs+7个随机残基+2-6c末端残基)。5个不同的c末端结尾在此列出:
seqidno01(wr),seqidno02(wlr),seqidno03(wqsr),seqidno04(wltvr),seqidno05(wqeggr)。
包含动态编码文库的nestlink表达载体plnx
nestlink表达载体plnx携带具有108种序列变异体的动态编码文库(图2),并使得在具有动态编码的框架中引入感兴趣的蛋白质文库(ploi)。由于两个文库(ploi和动态编码文库)彼此嵌套,此步骤的结果是“嵌套文库”。表达载体还允许限制性酶切介导的嵌套文库(与动态编码融合的ploi)解离,以致其可插入深测序质粒或可使用双链寡核苷酸(接头)进行直接illuminamiseq接头连接。注意ploi可为任何基因编码文库。
通过限制性酶切消化编码文库的源dna之后连接至表达载体而将ploi引入表达载体。本发明的发明人使用iis型限制性酶(sapi)用于此目的。源dna通常在噬菌体展示筛选之后由所获得的噬菌粒产生,包含sapi定向位点,使得ploi可在无pcr扩增的情况下亚克隆至nestlink表达载体中(此载体的说明见下文)。当ploi被插入时,其取代负筛选盒(ccdb),这大大改进插入步骤的效率。
通过凝血酶从ploi切除动态编码,并通过胰蛋白酶从动态编码去除his标记。这些裂解确保具有最佳质量、最佳疏水性以及最佳电荷的肽被分离用于质谱(见上文动态编码说明)。任何其他的蛋白酶组合也用于相同目的,这也是可想到的。
值得注意的是,动态编码的c末端精氨酸(r)起重要作用:首先,由于动态编码中赖氨酸或其他精氨酸被省略,它是动态编码仅有的带正电荷的氨基酸。为此,胰蛋白酶—正电荷之后裂解并因此认为相当非特异性的蛋白酶—可用于特别裂解精氨酸和his标记之间的肽键(动态编码带有his标记用于质谱分析会相当重,且his标记会在质谱之前减少反相色谱中的分离)。其次,已知具有c末端精氨酸的肽可通过质谱分析很好地检测到(有利的电离特性)。第三,因为此单正电荷氨基酸存在于动态编码中,总电荷始终为2+(n末端+精氨酸,在低检测ph所有其他残基为中性),这有利于数据分析。
此技术的重要方面是下述事实,即,可以或有必要将数种独特的动态编码连接至感兴趣的蛋白质文库的相同成员。例如,为了分析具有100种不同蛋白质的池,将2000个动态编码连接至这100种蛋白质,以致,所述此的各蛋白质与不同动态编码连接20次(池成员和动态编码之间的比率可实际上随需要而变化)。丰余标记有利于通过多个动态编码序列对池成员进行明确检测,并使动态编码序列对所分析的感兴趣的蛋白质的生物物理特性的潜在影响平均。丰余标记还使得能够确定所筛选的样品中不同蛋白质文库成员或不同筛选样品中相同蛋白质文库成员的相对定量。丰余还另外为技术原因所需:尽管动态编码被设计用于通过质谱分析的最佳检测,一些动态编码将检测不到,原因是它们在样品制备过程中丢失,或在反相柱的疏水性窗口中未洗脱,这通过质谱分析来分析。
另外,nestlink表达载体包含允许切离嵌套文库(与动态编码融合的ploi)的两个sfii限制性酶切位点,以致它可插入深测序质粒,或illuminamiseq接头可使用双链寡核苷酸(接头)直接连接。该关键步骤的原理在下文提供。
值得注意的是,ploi中或ploi和动态编码之间的sfii限制性酶切位点和/或其他限制性酶切位点可用于将另外的序列添加至嵌套文库。由此,这些另外的序列可作为与嵌套文库的融合序列来表达(在动态编码和ploi之间或邻接嵌套文库)。重要的是,由于在引入这些另外的序列之前进行向深测序质粒的转移(或通过寡核苷酸对接头连接直接进行深测序),这些序列不增加深测序阅读长度(由于技术原因是有限的)。另外,以此方式添加另外的序列保持动态编码和ploi之间的物理连接,这对于动态编码正确配置于ploi成员是绝对重要的。
深测序质粒
深测序质粒是一组携带用于通过illuminamiseq深测序的所有必要序列并允许插入来自nestlink表达载体的嵌套文库成员的池的载体。
将嵌套文库转移至深测序质粒(图1和2)通过限制性酶消化和连接而进行。本发明的发明人使用限制性酶sfii用于此目的,原因是其具有足够的高特异性,当消化可偶然编码限制性位点的整个文库时这是重要的。另外,所选择的sfii识别位点翻译成适当柔韧的和疏水性氨基酸,所述氨基酸可用作表达载体中的连接氨基酸。
本发明的发明人通过试验显示,对于nestlink来说,将nestlink表达载体转移至深测序质粒的步骤不包括嵌套文库的pcr扩增步骤。蛋白质—动态编码序列的pcr扩增不可避免地导致文库成员之间非同源性区(例如cdr)和动态编码(一种目的蛋白的动态编码非预期地连接至另一蛋白的动态编码,其中所述另一蛋白的动态编码不与nestlink表达载体连接)的重组。由此破坏动态编码和蛋白质之间的键合。
如上所述,通过sfii从表达载体切除嵌套文库。接下来,将其连接至深测序质粒中。其取代负筛选盒(ccdb),这对于插入步骤的效率是重要的。插入之后,其侧邻对于由illuminamiseq深测序所必要(和频繁使用)的测序。由朝向中心两侧开始测序。由此,相关区域以相对方向存在于插入片段两侧(除索引以外的反向互补序列)。
下述是对于所述序列从内部(插入片段)向外部区域的描述:
sfii位点:用于通过嵌套文库取代ccdb。
多样性:基于紧靠引物结合位点的序列,illuminamiseq技术生成第一测序信号。前面的少数碱基必须多样(不全同的),以防止检测通道信号超负荷和测序运行受破坏。
引物结合位点:测序引物在此结合。
索引(使用501和701号码标记):illuminamiseq技术允许多路技术,即刻在一个测序运行中分析数个样品。为了确保读数术语各样品,还对索引进行读数(可变的8bp伸展区)。为了在一个深测序运行中能够对数个nestlink试验进行测序,本发明的发明人生成了具有11种深测序质粒的组,各深测试质粒携带不同索引对(注意,插入片段两侧均有索引序列)。
接头:这用于固定dna模板以用于在illuminamiseq流式细胞上进行深测序。
bseri限制性酶切位点:这用于生成线性dna片段,这是illuminamiseq深测序所需要的。bseri是iis型限制性内切酶(切除其识别序列外部)的事实特别可用于使接头上的悬突最小化。
在传统方法中,通过pcr、通过illumina接头连接、之后pcr扩增、或者通过truseqdnapcr-freesampleprep试剂盒(illumina),将所有这些illuminamiseq序列元件连接至待测序的dna。在本发明的发明人的方案中,通过限制性酶切和连接从供体载体(本文nestlink表达载体)将待测序的dna(本文蛋白质—动态编码序列)亚克隆至深测序载体中,由此免除pcr。在最后的步骤中,使用bseri裂解深测序载体。这释放完整的illuminamiseq测序模板,所述模板通过dna琼脂糖凝胶从载体骨架分离,并通过凝胶提取纯化。
用于深测序的双链接头—寡核苷酸
允许将illuminamiseq深测序所需接头序列pcr非依赖性连接至ploi的第二个策略依靠携带与所述深测序质粒相同一组接头序列的双链寡核苷酸,所述双链寡核苷酸可通过基因合成互补单链寡核苷酸和接着退火反应而生成。合成长度不同的互补单链,产生退火接头的粘性悬突。此悬突对应于sfii限制性酶切位点的互补序列,当从nestlink表达载体切除动态编码ploi时而产生。因此,可将退火的寡核苷酸与动态编码ploi高效率连接以连接illuminamiseq深测序所需的接头序列。在深测序之前通过琼脂糖凝胶纯化连接产物。
如下述从内部(插入片段)向外部区域描述最终深测序模板的序列:
动态编码ploi:通过sfii限制性酶消化从nestlink表达载体切除动态编码ploi。
sfii限制性酶切位点的剩余部分:此酶允许从nestlink表达载体切除,所产生的粘性末端用于位点特异性连接深测序接头。
多样性:基于紧靠引物结合位点的序列,illuminamiseq技术产生第一测序信号。前面的少数碱基必须多样(不全同的),以防止检测通道信号超负荷和测序运行受破坏。
引物结合位点:测序引物在此结合。
索引(使用501和701号码标记):illuminamiseq技术允许多路技术,即刻在一个测序运行中分析数个样品。为了确保读数术语各样品,还对索引进行读数(可变的8bp伸展区)。为了在一个深测序运行中能够对数个nestlink试验进行测序,本发明的发明人生成了具有7种深测序质粒的组,各深测试质粒携带不同索引对(3种用于一个末端,4种用于另一末端),这允许生成12种不同索引对。
接头:这用于固定dna模板以用于在illuminamiseq流式细胞上进行深测序。
通过动态编码对ploi成员进行定量
许多nestlink应用要求对动态编码ploi成员进行绝对定量。尽管lc-ms对于蛋白组学中各肽的定量不够精确,但nestlink受益于与各ploi成员连接的多个动态编码,并受益于同质动态编码文库,所述同质动态编码文库被设计用于通过质谱分析进行最佳检测。基于此考虑,本发明的发明人推定任何给定的ploi成员的所有动态编码的ms1强度总和必须与样品中此ploi成员的定量成比例。本发明的发明人通过将已知量的与不同数量动态编码连接的八种合成抗体掺加为分别包含来自大肠杆菌和耻垢分枝杆菌的裂解物的两种样品(图3)验证了此推定。所观察到的各动态编码合成抗体的所有动态编码的ms1强度总和之间的线性关系及其掺加定量证实了此推定的正确性,并证实本文描述的nestlink过程可用于对池中各ploi成员进行定量。如果在用于lc-ms的动态编码分离之前已知量的一个或多个动态编码蛋白质(标准)被掺加至样品,则可确定各ploi成员的绝对定量。
用于噬菌体展示筛选的噬菌粒(nestlink前)
在本发明的发明人当前的应用中,ploi是富集的合成纳米抗体的池。通常,大的合成抗体文库使用噬菌体展示富集以结合靶蛋白。为了避免非同源性区(即cdr)重组,在噬菌体展示筛选之后,ploi必须不被pcr扩增。为此目的,构建噬菌粒载体(图2a),使得可通过sapi限制性酶切位点将ploi亚克隆至nestlink表达载体中。值得注意的是,sapi位点是翻译产物的一部分,在噬菌体表面展示。本发明的发明人可试验性显示这些来自sapi位点的另外的氨基酸茎不干扰噬菌体展示效率。
除了sapi位点以外,噬菌体展示载体包含通常存在于噬菌粒中的所有用于在m13噬菌体上展示蛋白质的元件,且是载体pmesy4(genbankkf415192)的衍生物。
另外一个对于本文所述的所有载体相关的通常注意点:为了使得将插入片段有效从一个载体转移至另一载体,重要的是所述载体携带不同抗生素抗性。因此,nestlink表达载体携带氯霉素抗性标记,深测序载体携带卡那霉素抗性标记。另外,用于噬菌体展示筛选的噬菌粒包含氨苄青霉素抗性标记。
原理证明试验
在此试验中,本发明的发明人证明nestlink可用于以非先验方式表征蛋白质候选物大池中各蛋白质,并证明可鉴定具有用于下游选择的应用最有前景的特性的池成员。
更特别地,下文描述的原理证明试验表明:i)开发了一种用于在良好控制的文库多样性下进行文库嵌套的有效防范;和ii)所嵌套的文库可用作结合蛋白池无先验筛选压力的基础。
在此实施例中,本发明的发明人使用由合成抗体池组成的ploi进行工作,所述ploi通过核糖体和噬菌体展示(未描述)针对麦芽糖结合蛋白(mbp)预富集。
本发明的发明人使用此专利中描述的方法一次将下述筛选压力施用于合成抗体的多样性池:i)筛选最高表达的合成抗体;ii)筛选具有最高可溶性的合成抗体;以及iii)在溶液结合测定中筛选结合靶的合成抗体。
使用材料和方法部分描述的方案,本发明的发明人意于将约1200个不同合成抗体池成员连接于约17’000种独特的动态编码,导致所谓的“嵌套文库”。这通过首先在一个容器中培养包含编码合成抗体的噬菌粒的一定合适克隆数量的细胞之后分离其质粒dna来实施。在转化后通过铺于琼脂糖板而非单个挑选合成抗体克隆来估计每一体积回收细菌的集落形成单位数量(cfu)。因此,合适体积的回收细菌(约1’200cfu)用于培养物接种,所述培养物之后被收获用于质粒dna分离。然后,将这些多样性限制的噬菌粒的dna插入片段与包含具有约108个不同变异体的动态编码文库的表达载体pnlx连接。使用如上所述的cfu估测,将克隆数量限制至约17’000。由于仅使用108个变异体中的约17’000个包含动态编码的载体(如通过cfu估测所确定的),本发明的发明人计算出99.974%的动态编码是独特的,因此,一种独特合成抗体上标记有绝大多数动态编码。另外,由于它们嵌套约17000个包含动态编码的载体中的约1’200个合成抗体基因,预期平均每个合成抗体使用14种不同动态编码标记。
载体pnlx中的嵌套文库在单个烧瓶的细菌中表达,并作为动态编码结合蛋白池纯化以进行筛选试验(见下文)。为了对嵌套文库进行测序,将动态编码合成抗体转移至深测序载体pnl,所述深测序载体pnl携带所有使用miseq装置进行illumina深测序的相关序列。嵌套文库的深测序使得每一动态编码明确配置于其相应的合成抗体。深测序数据与嵌套文库中预期的合成抗体和动态编码数量一致,原因是在数据过滤之后,获得与13’620种独特的动态编码连接的1080个不同合成抗体序列。因此,平均每个合成抗体与不同动态编码连接12.61次。测序数据过滤之后,本发明的发明人未观察到不明确的动态编码与合成抗体的连接(即相同动态编码与两种或多种不同合成抗体连接)。根据本发明发明人的知识,这种使用良好控制的多样性在彼此之间嵌套文库的成功尝试是无先例的。
使用深测序数据,通过将各合成抗体的所有动态编码连锁至理论连续的蛋白质序列(相应的合成抗体作为鉴定剂),构建携带嵌套文库完整序列信息的数据库。然后将此数据库上载于mascot服务器,之后用于ms/ms离子检索。
作为此技术的新型应用的实例,本发明的发明人使用嵌套文库,特别筛选和鉴定具有一定表观流体力学半径的那些合成抗体,和在溶液中表现与mbp高亲和力相互作用的那些合成抗体。这些特性均通过尺寸排阻色谱(sec)测定,使用需要基因型—表型连锁的当前现有技术展示系统不具有顺从性,原因是所述基因型通常增加所展示的蛋白质的大小100多倍,致使展示颗粒对蛋白质水平的小尺寸差异不灵敏。
为此,表达嵌套文库,通过ni-nta树脂纯化动态编码结合蛋白,并使其经受sec。汇集对应于单体蛋白质的(具有最高可溶性的结合蛋白候选物)合成抗体的洗脱级分,将其分成两个等量的整分试样。一个整分试样与mbp一起温育,另一整分试样仅与缓冲液一起温育。在sec上分离分析两个样品(无mbp的运行用作对照),收集对应于合成抗体—mbp复合物尺寸的洗脱级分。接下来分离所收集的mbp级分和对照运行的动态编码,在同量异序标记对分离的动态编码标记之后,使其经受两个分离的lc-ms运行,或合并至一个lc-ms/ms运行。然后,先前生成的深测序数据库(动态编码为合成抗体配置)可在masot检索中用于鉴定动态编码,由此明确鉴定在合成抗体-mbp复合物尺寸洗脱的合成抗体。此试验使得本发明的发明人鉴定了超过300种独特的合成抗体,所述独特的合成抗体均良好表达、为单体并结合溶液中的靶蛋白质。
将nestlink用于解离速率测定
为了对在上述原理证明试验中鉴定的mbp特异性合成抗体进行评分,本发明的发明人通过生物素化mbp将等量分离的mbp合成抗体复合物固定于两个链霉亲和素—琼脂糖凝胶柱(图4)。然后在一个柱子上使用过量非生物素化mbp(洗涤3分钟)进行解离速率筛选,而另一个柱子仅通过缓冲液洗涤。洗涤之后,洗脱来自两个柱子的剩余的合成抗体,使其动态编码经受两轮lc-ms/ms。与上述溶液中结合试验(sec运行)相似,mascot检索中使用深测序数据库用于通过动态编码进行合成抗体鉴定。另外,使用progenesis软件对各合成抗体总计所有鉴定的动态编码的ms1强度。由于如上所确定的ms1峰强度的定量性质,本发明的发明人预期两个柱子的各合成抗体的动态编码—强度—总和之间的比率对应于使用过量非生物素化靶进行解离速率筛选之前和之后其相对浓度。推定单指数衰减之后是各解离反应,和使用关于以过量靶洗涤时间的知识(3分钟),由此作者能够一次确定超过300种结合蛋白的大致解离速率。此分析通过使用表面等离振子共振检测11种结合蛋白各自的解离速率来证实。根据作者的知识,在单个试验中确定结合蛋白候选物池中的解离速率是无先例的。先前由于要求处理各蛋白质而需要数周的过程现在可使用本文描述的技术一次实施。
将nestlink用于来自免疫的驼科的结合蛋白鉴定
将nestlink用于天然纳米抗体池,所述天然纳米抗体池通过从免疫的羊驼(驼科)的b细胞进行cdna分离而获得。用于免疫的抗原是tm287/288,一种来自海栖热袍菌(thermotogamaritima)的abc运载蛋白(膜内在蛋白)。与传统的从驼科生成纳米抗体相对,此纳米抗体池并非使用噬菌体展示针对靶而富集。
将所述纳米抗体pcr扩增、多样性限制以及与动态编码文库交错而产生3’469种独特的纳米序列,如通过illuminamiseq深测序所确定的,所述纳米序列与59’974种独特的动态编码连接(见材料和方法部分)。通过ni-nta表达和纯化嵌套文库,之后通过sec分离单体池成员。与原理证明试验(上述)类似,在此预筛选步骤中去除不表达或不可溶的不利的结合蛋白候选物。在从ni-nta柱和sec运行的单体级分洗脱之后,收集lc-ms样品。接下来,将增加量的池与tm287/288以约0.1:1、2:1以及100:1的比率一起温育,使抗原/池混合物再经受三轮sec(图5)。收集对应于靶/纳米抗体复合物尺寸的级分。通过lc-ms/ms分别分离和分析所有收集的样品的动态编码,这允许一次对所有结合蛋白比较表达水平、可溶性(sec上的单体)以及对溶液中抗原的结合强度。
在此对于来自免疫的驼科的3’469种独特的纳米抗体的分析中,本发明的发明人鉴定了27种具有有利稳定性、表达水平以及可溶性的高亲和力结合蛋白家族。值得注意的是,nestlink比使用elisa和sanger测序的噬菌体展示筛选和过量传统筛选有效的多,后者在显著较长的处理时间内于仅鉴定相同池中这些家族中的5个家族。总而言之,因此可以说明,nestlink可用于从免疫的驼科鉴定最有前景的候选生物分子,其具有当前现有技术方法满足不了的处理量和精确度。
将nestlink用于鉴定靶向细胞表面的蛋白质的结合蛋白
以鉴定溶液中针对纯化的靶/抗原的结合蛋白的目标进行上述试验,其产生用于体外应用(例如晶体学)的有利研究工具。本文中,本发明的发明人意于解决药物开发的核心薄弱环节,即,鉴定以高特异性和亲和力识别细胞表面靶蛋白的膜蛋白结合蛋白。开发针对膜蛋白靶的生物分子药物通常需要两个连续基本不同的步骤。首先,通过展示方法或免疫生成结合蛋白候选物的多样性池。其次,在细胞测定中对多样性池筛选结合和功能。后者固有地无效和缓慢,原因是其需要逐个分析各结合蛋白候选物(荣成以小型模式)。在此试验中,本发明的发明人通过nestlink取代第二个(筛选)步骤,以鉴定针对膜内在蛋白具有特异性的细胞表面结合蛋白,不需要对各结合蛋白候选物逐个进行费力的分析。
首先,本发明的发明人针对革兰氏阴性细菌的去垢剂溶解的纯外膜蛋白抗原进行合成蛋白文库的体外展示(步骤1,生成结合蛋白候选物的多样性池)。取代检测此多样性池各结合蛋白候选物的细胞表面结合(通常步骤2),本发明的发明人一次实施nestlink和检测大的候选物池(图6a)。将1’456种合成抗体与动态编码文库交错,产生31’500种动态编码的连锁(平均22种动态编码/合成抗体)。如上所述,通过深测序获得动态编码至结合蛋白的配置,表达、纯化嵌套文库,分离单体池成员(计数筛选/去除不想要的结合蛋白候选物)。因此,首先去除具有不良表达水平和不良可溶性的池成员,监测各池成员的表达水平和可溶性特性。由此,nestlink程序将具有前景的结合蛋白候选物集中于细胞表面筛选,这如下所述进行:将单体池成员分成4个当量的级分,将各级分与另一菌株一起温育。通过沉淀和使用缓冲液重悬/洗涤去除未结合的合成抗体候选物。接下来,分离与其中一种菌株结合的合成抗体的所有动态编码,使其经受lc-ms分析。每一合成抗体的所有动态编码的所有ms1强度总和用作池中各靶细胞的各合成抗体的相对浓度的测量值。这使得精确读出细胞特异性(图6)。
从池中1’456种结合蛋白候选物,鉴定6种良好表达和可溶的合成抗体,所述合成抗体特异性识别以天然形式包埋在革兰氏阴性细菌(菌株4)外膜中的蛋白靶。本发明的发明人通过针对4种菌株的流式细胞术各自分析这6种鉴定的合成抗体验证了此发现(在荧光标记所述合成抗体之后)。如通过nestlink所观察到的,在此单克隆测定中所有检测的候选物均表现相同的特异性模式。值得注意的是,如通过illuminamiseq深测序所测定的,各鉴定的结合蛋白在嵌套池中仅以<0.05%存在。考虑到现有技术筛选仅考虑结合蛋白候选物的一个特性(例如靶结合),在报告表达水平或可溶性/寡聚化倾向方面具有缺陷,这6种结合蛋白中的任何结合蛋白均可通过传统单克隆筛选方法鉴定是不可能的。由此,此试验证实了,由于缺乏基因型-表型连锁和两个文库的交错,nestlink允许以无先例的深度筛选结合蛋白文库。
将nestlink用于监测模型生物中生物分布和药物代谢动力学参数
在先前的实施例中,本发明的发明人显示,由于缺乏基因型—表型连锁,nestlink筛选允许无先例筛选过程(例如在sec上筛选单体池/文库成员)。本文中,引入在物理基因型—表型连锁情况下不能实现的另一筛选压力,筛选活生物体中具有特定生物分布和药物代理动力学特性的蛋白质。可将治疗性生物分子候选物的嵌套(动态编码标记的)池注射至动物模型中,可在一定实耗时间后通过lc-ms检测机体内各部位(例如,不同器官、组织或肿瘤等)各池成员的相对浓度。此类型分析导致在特定时间点对于机体内各池成员进行综合/总体生物分布分析。如果相同物种的数种类似单体在不同时间点之后经受此分析,nestlink生物分布分析可被扩展为时间维度,由此允许在低度或中度时间分辨率对于各候选物进行药物代谢动力学数据获取。
本发明的发明人从先前使用不同数量动态编码合成抗体掺加的匀化小鼠组织通过检测和最优化动态编码提取过程而为此类型分析设定基础。详细地,首先将数种合成与小量动态编码(20-30)的连接,通过illuminamiseq深测序确定合成抗体至动态编码的配置。然后,动态编码标记的合成抗体各自表达和纯化,通过吸光度测量来测定。然后以不同浓度(跨一个数量级)合并各合成抗体。
平行地,融解冷冻的小鼠器官(肝脏、肺脏、肾脏)以及血液,并使用变性缓冲液条件和陶器匀化。将先前制备的滴定混合物掺加为匀浆,在室温下温育30分钟,以使得潜在的蛋白酶或动态编码修饰的酶起作用。接下来,提取合成抗体连同剩余的其动态编码,通过蛋白酶裂解分离动态编码,并通过lc-ms分析。基于各滴定混合物的合成抗体的检测,本发明的发明人发现,通过lc-ms从所述匀化器官和组织进行合成抗体检测通常依赖于30–100ng的定量(合成抗体)。考虑到达1mg的治疗剂通常可注射至小鼠模型中,明确的是,在注射嵌套池之后,在机体的大多数相关部位会存在数个微生物。由此,存在足够的非降解和非修饰动态编码以监测结合蛋白池的总体生物分布,并进行其药物代谢动力学分析。
材料和方法
在下述内容中,提供了nestlind方法的通用方案。其包含上述所有用于实施试验的步骤,并提供了涉及动态编码结合蛋白池的文库嵌套、深测序、表达以及纯化、动态编码提取、lc-ms以及数据分析的细节。
通过文库嵌套对动态编码纳米抗体进行克隆
1.合成抗体/纳米抗体池的多样性限制
已使用合成抗体池或天然纳米抗体进行nestlink试验,所述合成抗体池或天然纳米抗体分别通过噬菌体展示或免疫从体外结合蛋白筛选而获得。在噬菌体展示用于结合蛋白筛选的情况中,将200ng体外筛选的在噬菌粒中编码的潜在结合蛋白池转化至50µl大肠杆菌mc1061化学感受态细胞(通过promegacorporation,subcloningnotebook2004的方案实现感受态)。将稀释系列铺于包含120µg/ml氨苄青霉素的琼脂糖凝胶培养板,并在30°c过夜温育。通过2ml的包含100µg/ml氨苄青霉素的lb培养基重悬浮包含所需集落形成单位(在上述实施例中,数量范围为1000cfu和1500cfu之间)的培养板的集落,将悬浮液转移至200ml包含100µg/ml氨苄青霉素的lb培养基的培养物。在37°c过夜培养此培养物,将其用于dna制备(试剂盒:#740412.10,macherey-nagel)。在50°c、140µl反应体积中通过于缓冲液neb3.1(newenglandbiolabs,#b7203s)中的100单位的bspqi(newenglandbiolabs,#r0712l)消化15µg制备的噬菌粒1小时,之后在80°c灭活酶20分钟。在2%(w/v)琼脂糖凝胶上进行电泳,切除并提取对应于结合蛋白池的条带(试剂盒:#740609.250,machery-nagel)。在免疫的羊驼的情况中,如所述从b细胞的cdna扩增纳米序列(pardon等,natprotoc.,2014mar;9(3):674-93),使用包含bspqi限制性酶切位点的引物扩增。在50°c、140µl反应体积中通过于缓冲液neb3.1(newenglandbiolabs,#b7203s)中的100单位的bspqi(newenglandbiolabs,#r0712l)消化5µg纯化的pcr产物1小时,之后在80°c热灭活酶20分钟。在2%(w/v)琼脂糖凝胶上进行电泳,切除并提取对应于结合蛋白池的条带(试剂盒:#740609.250,machery-nagel)。将消化的pcr片段克隆至具有卡那霉素抗性标记的fx克隆起始载体中(geertsma等,biochemistry,2011apr19;50(15):3272-8),通过2ml包含50µg/ml卡那霉素的lb培养基重悬浮3.500cfu,将悬浮液转移至200ml包含50µg/ml卡那霉素的lb培养基的培养物。在37°c过夜培养此培养物,将其用于dna制备(试剂盒:#740412.10,macherey-nagel)。在50°c、140µl反应体积中通过于缓冲液neb3.1(newenglandbiolabs,#b7203s)中的100单位的bspqi(newenglandbiolabs,#r0712l)消化15µg制备的噬菌粒1小时,之后在80°c热灭活酶20分钟。在2%(w/v)琼脂糖凝胶上进行电泳,切除并提取对应于结合蛋白池的条带(试剂盒:#740609.250,machery-nagel)。
2.将动态编码连接于合成抗体/纳米抗体池和动态编码多样性限制
如上所述通过bspq1消化包含动态编码文库的载体pnlx用于噬菌粒,在1%(w/v)琼脂糖凝胶上进行电泳。,切除并提取对应于开放载体的条带(试剂盒:#740609.250,machery-nagel)。在37°c、28µl反应体积中,使用于t4连接酶缓冲液(fermentas#b69)中2.5单位的t4连接酶(fermentas#el0011)将200ng结合蛋白池连接于400ng消化的pnlx,之后在65°c热灭活10分钟。将25µl连接反应液用于转化至150ul电感受态大肠杆菌mc1061细胞(根据howard和kaser2007,makingandusingantibodies,第170页制备)。在soc培养基中于37°c恢复细胞30分钟,使用一定体积恢复的细菌接种200ml包含25µg/ml氯霉素的培养物,所述一定体积恢复的细菌对应于通过将稀释样品铺于包含25µg/ml氯霉素的琼脂糖培养板所测定的所需数量集落形成单位(在上述实施例中,cfu数量范围在13’000和30’000之间)。在37°c过夜培养培养物,之后进行dna制备(试剂盒:#740412.10,macherey-nagel),生成包含与1ml的50%(v/v)甘油混合的1ml静止期培养物的甘油原种。
深测序
1.illumina接头序列的连接
在50°c、140ul反应体积中,通过于缓冲液g(fermentas#bg5)中120单位sfii(fermentas#er1821)消化15µg的包含动态编码结合蛋白的pnlx,之后添加12µl的0.5medta用于酶灭活。在2%琼脂糖凝胶上进行电泳,切除并提取对应于与动态编码连接的结合蛋白池的条带(试剂盒:#740609.250,machery-nagel)。对于第一个使用抗mbp的合成抗体的实例,如上对于pnlx所述,通过sfii消化包含接头的载体pnl,所述接头为使用合适索引通过illuminamiseq进行dna深测序相关的(在此情况,502和703用于双索引),于1%琼脂糖凝胶进行电泳。切除并提取对应于载体骨架的条带(试剂盒:#740609.250,machery-nagel)。在37°c、28µl反应体积中,使用于t4连接酶缓冲液(fermentas#b69)中2.5单位的t4连接酶(fermentas#el0011)将400ng动态编码结合蛋白池连接于300ng消化的pnlx,之后在65°c热灭活10分钟。将25µl连接反应液用于转化至250ul电感受态大肠杆菌mc1061细胞(根据howard和kaser2007,makingandusingantibodies,第170页制备)。在soc培养基中于37°c恢复细胞45分钟,使用所有恢复细胞接种200ml包含30µg/ml卡那霉素的培养物。将试验样品铺于卡那霉素选择性琼脂糖培养板,以验证连接和转化效率足以转移完整嵌套文库(总共>200’000cfu)。在37°c过夜培养培养物,之后进行dna制备(试剂盒:#27106,quiagen)。在37°c、20µl总反应体积中,使用于cutsmart缓冲液(newenglandbiolabs,#b7204s)中的5单位的bseri(newenglandbiolabs,#r0581s)对1µg包含动态编码结合蛋白池的制备的pnl进行限制性酶消化2小时,之后在80°c灭活酶20分钟。注意,此时可汇集针对不同靶的数个动态编码池(在bseri消化之前),各动态编码池位于不同索引的pnl。接下来从1%琼脂糖凝胶提取包含与miseq接头连接的动态编码结合蛋白池的插入片段。
对于上述提供的其他实施例,在37°c、20µl反应体积中,使用于t4连接酶缓冲液(fermentas#b69)中5单位的t4连接酶(fermentas#el0011)将300-400ng退火的包含粘性sfii悬突的寡核苷酸与600ng通过sfii从pnlx切除的动态编码结合蛋白池混合,之后在65°c热灭活10分钟。接下来从2%琼脂糖凝胶提取与miseq接头连接的动态编码结合蛋白池(试剂盒:#740609.250,machery-nagel)。注意,此时可汇集针对不同靶的数个动态编码池,各动态编码池包含不同的连接的接头对。
2.纳米抗体—动态编码连锁的测定
在来自illumina的miseq装置上使用双末端对方案进行深测序(miseq试剂盒v2(300个循环))。在分析的第一个步骤中,使用标准软件(illumina)将双末端读数缝接在一起。对于任何给定的索引对,总共获得800’000–8百万的读数,这对应于25-70的平均读数冗余(此数字等于通过给定嵌套文库的总预期动态编码数除总读数)。使用定做的脚步,通过应用下述阳性标准过滤所产生的原始读数:i)动态编码不变部分的正确侧邻模式,ii)纳米抗体不变部分的正确侧邻模式,iii)序列不包含n,iv)序列位于可能的纳米抗体—动态编码融合物的预期尺寸范围内,v)纳米抗体—动态编码融合物的序列位于框内(即刻被3除),vi)序列无终止密码子。在过滤之后,生成一系列独特的动态编码。被读取至少5次的动态编码被认为是正确的。对于各正确的动态编码,生成所有连接的纳米抗体序列的共有序列。需要共有序列方法来校正纳米抗体序列中的测序错误。共有序列评分被引入以监测与相同动态编码连接的纳米抗体序列中的可变性。在一个或数个与相同动态编码连接的纳米抗体明确相互不同的情况,所述评分给出大的罚分,由此去除与两个或多个不同纳米抗体连接的动态编码用于进一步分析。仅具有高共有序列评分的纳米抗体—动态编码对被进一步考虑。在最后的步骤中,鉴定相同的(共有)纳米抗体序列,及其所有连接的动态编码(在上述实施例中,平均每个纳米抗体12-40个动态编码)。使用纳米抗体序列作为鉴定剂将所有与相同纳米抗体连接的动态编码均串接至推定的蛋白质序列中,以fasta文本格式保存此数据库。
单体动态编码合成抗体/纳米抗体的表达和纯化
包含携带动态编码结合蛋白池的pnlx的大肠杆菌mc1061甘油原种用于接种包含50ml包含1%葡萄糖的lb预培养物,所述预培养物在37°c过夜培养。通过预培养将600mltb培养物接种至0.05的od,在37°c培养1.5小时,之后在20°c过夜培养。在0.8的od600通过0.05%(w/v)阿拉伯糖进行诱导。通过在5’000g旋转20分钟收获细胞。倒出上清液,将细胞重悬于25ml补充有一捏dna酶i(sigma#dn25)的50mmph7.5tris-hcl(20°c),150mmnacl,15mmph8.0咪唑(20°c)中。使用微流化仪(microfluidics#11op)在30’000psi裂解两轮,同时在冰上冷却。在5’000g沉淀细胞碎片30分钟,通过重力流动将上清液应用于1.5mlni-nta超流柱(quiagen#1018142)。通过30ml包含20mmph7.5tris-hcl(20°c)、150mmnacl以及300mmph8咪唑(20°c)的洗涤缓冲液洗涤柱子。将5ml洗脱液注射至hiload16/600superdex200pg(gehealthcarelifesciences#28989335),收集对应于单体级分的区域,在nanodrop2000c(thermoscientific)于2.1吸光度(280nm)针对缓冲液浓缩至1.2ml体积,用于上述实施例所列的进一步的筛选试验。
动态编码的分离
通过ex缓冲液(20mmtris-hclph8.5,150mmnacl,0.5%(v/v)tritonx-100,0.125%(w/v)去氧胆酸钠,10mmph8.0咪唑,4.5mgdmcl)洗脱包含动态编码ploi的样品10-20次,过滤(注射式过滤器,0.2µm截断率),在室温下将其与100ulni-nta超流淤泥(quiagen#1018142一起温育2小时。接下来,在500g沉淀树脂10分钟,将其转移至小生物旋转色谱柱,之后使用ex缓冲液3x500µl洗涤,使用包含30mmph8.0咪唑的th缓冲液(20mmteabph8.0,150mmnacl,2.5mmcacl2)3x500µl洗涤,以及使用th缓冲液3x500µl洗涤。在封闭柱子底端之后,将树脂重悬浮于100µl包含2.4u凝血酶(millipore#69671-3)的th缓冲液中,之后在室温下过夜温育。然后,通过3x500µl包含30mmph8.0咪唑的th缓冲液对柱子引流和洗涤,之后通过3x500µltry缓冲液(20mmph8.0teab,50mmnacl,2.5mmcacl2)引流和洗涤,通过包含300mmph8.0咪唑的try缓冲液洗脱。通过预平衡(h2o)的microcon10kda截断浓缩器(amicon:ym-10)旋转洗脱物,将1µg胰蛋白酶(promega#v5113)添加至滤液中,之后在37°c过夜温育。
接下来,使洗脱的动态编码经受ziptip(millipore#ztc18s960)净化程序。通过200µl甲醇、200µl60%(v/v)乙腈(acn)以及200µl包含0.1%(v/v)三氟乙酸的3%(v/v)乙腈预洗涤ziptip。上载100µl动态编码样品,之后使用200µl包含0.1%(v/v)三氟乙酸的3%(v/v)乙腈预洗涤,通过2x40µl的包含0.1%(v/v)三氟乙酸的60%(v/v)乙腈洗脱。接下来,蒸发溶剂(真空离心),将动态编码重悬浮于15µl包含0.1%(v/v)甲酸的3%(v/v)乙腈。
lc-ms
使用easy-nlc1000hplc系统,将2µl重悬浮的动态编码溶液注射至填充有反相材料的内部制备的毛细管柱(reprosil-pur120c18-aq,1.9µm;柱尺寸150mmx0.075mm)。使用溶剂a(于水中的0.1%甲酸(fa))平衡柱子。使用下述梯度以0.3µl/分钟的流速洗脱肽:0-60分钟;5-20%b(于acn中的0.1%fa),60-70分钟;20-97%b。在通过97%b洗涤10分钟之后,通过溶剂a再平衡柱子5分钟。使用orbitrapfusion质谱仪利用下述参数获取噶精确性的质谱:300-1500m/z的扫描范围,5e5的agc-靶,120’000的分辨率(m/z190),以及100ms的最大注射时间。使用四极分离(1.6m/z窗口)、1e4的agc靶,35ms最大注射涉及,以30%碰撞能进行的hcd裂解、3秒钟的最大循环时间在线性离子阱中以最高速度模式记录数据依赖性ms/ms波谱,所有可平行化的时间。筛选单同位素前体信号用于以2和6之间的电荷状态和5e4的最小信号强度进行ms/ms。将动力学排斥设置为25秒钟,和10ppm的排斥窗口。数据收集之后,使用proteomediscoverer1.4(thermoscientific)生成峰系列。
数据分析和定量
通过软件xcalibur预检查lc-ms运行(每动态编码提取物/样品一次运行),输入xcalibur原始文本,通过progenesis转化为mznld文本。接下来,progenesis用于对齐感兴趣的lc-ms运行(对齐分数>80%),从分析中去除带+1和+5至+20电荷的肽离子。接下来,从progenesis输出所有对齐的lc-ms运行的合并mgf文本(列阈值<5,离子片段计数>1’000,去同位素和电荷重叠合法),将其与先前测定的至ploi成员配置的动态编码(fasta文本格式的深测序数据库,见上文)一起上载于mascot服务器。将mascot识别直接输入scaffold软件,之后进行数据转化,输出波谱报告,接下来将其输入progenesis,这允许将特性配置于其对应的动态编码。使用progenesis,通常将特性强度标准化为掺加的标准,各ploi成员的所有独特动态编码用于定量。接下来,输出原始和标准化强度(csv格式),并通过excel进一步分析。
- 还没有人留言评论。精彩留言会获得点赞!