一种耐酸性光控荧光分子开关及其合成方法和应用与流程
本发明属于分子开关领域,具体涉及一种耐酸性光控荧光分子开关及其合成方法和应用。
背景技术:
近年来发展出一系列超高分辨率荧光成像技术,其中基于单分子定位的光激活定位显微技术(plam)和随机光学重构显微技术(storm或dstorm)使光学显微镜的空间分辨达到了前所未有的高度(20nm)。目前超分辨显微成像技术已经被广泛应用到生命科学研究中,然而尽管超分辨显微成像技术取得了巨大的进步,将荧光显微镜的空间分辨率推进到了20纳米,但是超分辨荧光显微成像技术仍然面临诸多技术问题,其中之一就是所必需的荧光染料性能亟需改善。基于单分子定位的超分辨显微成像技术需要荧光染料不仅满足光稳定性好和荧光亮度高的需要,还要具有光控荧光分子“开-关”功能,这样才能够实现单分子的定位和检测。因此开发高荧光强度和光稳定性,具有光控荧光分子开关性能的新型荧光染料是超分辨荧光成像的迫切需求和当前热点。
开发生物成像用单分子定位超分辨荧光染料,目前最好的方法是在高荧光强度和光稳定性的染料中引入光控分子开关。罗丹明类染料由于其突出的光性能,是目前超分辨中用的最多的一类染料。罗丹明染料的荧光分子开关“明-暗”状态是基于酰胺螺环的光控开关,即传统罗丹明螺酰胺在紫外光辐照下,会由不发光的闭环结构变为强荧光发射的开环结构。s.w.hell等人最早利用这一独特的光控分子开关将罗丹明螺酰胺标记在固定的ptk2细胞的微丝骨架上,利用单分子定位技术实现了超分辨成像。但是,包括罗丹明螺酰胺在内,光控开关分子在细胞内应用所面临的共同难题是需要紫外光作为“开-关”激活光,例如罗丹明螺酰胺需要用波长小于375nm的光来将闭环结构打开变为有荧光的开环结构,而紫外光会对细胞产生严重的光毒性,难以在活细胞中应用。为了改善激活光波长,w.e.moerner等人将酰胺取代基修饰为较大的共轭体系,将吸收波长向长波长移动,首次将开关激发光延长到可见光区(>400nm),实现了对细菌表面的三维超分辨荧光成像。由此,罗丹明螺酰胺在超分辨成像中表现出巨大潜力。
罗丹明螺酰胺实现了用可见光控制荧光分子“开-关”,这对于生物成像来说极其重要和有意义,但是这类分子依然存在一个严重缺点限制其在超分辨成像中的普遍应用,那就是罗丹明螺酰胺这种分子开关除了可以光控之外,还可以由酸碱来控制,即在酸性条件下,罗丹明酰胺螺环打开变成可发光的开环结构。而细胞内存在许多偏酸性的环境(如溶酶体,蛋白的酸性位点等),当罗丹明螺酰胺染料应用在这些酸性环境中,其酸控制的开关会严重干扰甚至导致光控分子开关性能完全失效,因此在酸性环境中基于这类染料的荧光探针目前无法应用于超分辨荧光成像。综上所述,开发耐酸性同时具有可见光控制的的罗丹明螺酰胺类荧光分子开关对于活细胞超分辨荧光成像显得尤为迫切和重要。
技术实现要素:
本发明提供了一种耐酸性光控荧光分子开关及其合成方法和应用,该分子开关是以3-伯胺或仲胺取代罗丹明螺酰胺染料为结构单元,研究发现这类开关染料在体内和体外的酸性环境下化学稳定。对3-伯胺或仲胺取代罗丹明螺酰胺进一步共轭修饰实现可见光(>400nm)激活螺环开关。将琥珀酰亚胺(nhs)活性酯和苄基鸟嘌呤(bg)引入罗丹明螺酰胺,这可以将荧光开关探针非特异性或特异性标记到生物蛋白上,通过storm技术就可以对目标蛋白进行超分辨成像。
本发明所述的一种耐酸性光控荧光分子开关,该光控荧光分子开关化学结构特征如下:荧光团母体是罗丹明类染料,罗丹明发色团母体存在不同的取代基r1、r2、r3、r4、x、y,内酰胺的邻位存在伯胺或仲胺取代基(即下式示中苯环的3位),其结构式如下所示:
其中r1、r2、r3、r4、r5是相同或不同的基团,为h、cmh2m+1、cmh2m-1、cmh2m-3、c6+mh5+2m、cmh2m+1co、cmh2m+1so2、cmh2m+1phso2中的任一基团;
x为o、c、si、ge、s、so2基团;
y为h、so3na;
z是cmh2m+1、cmh2m-1、cmh2m-3、c6+mh5+2m、(c2h4o)mch3、(c2h4o)moh、cmh2m(c4h8no)、6-(4-苯乙炔基)萘酐、6-(4-苯乙炔基)萘酰亚胺、4-苯乙烯基吡啶盐、6-(4-苯乙炔基)苝酰亚胺、或其带有单个或多个二级取代基团的衍生结构;
其中,二级取代基团是f、cl、br、i、r6、ca0a13、ca0a1a2a3、no2、or6、sr6、so2r6、sor6、so3r6、nhr6、nr6r7、cho、ch2or6、co2r6、ocor6、ococh2r6、chbcho、cb2cho、chbco2r6、chbor6中任何一种基团;
a0、a1、a2、a3和b是相同或不同的基团,为h、f、cl、br、i、no2、or6、sr6、nhr6、nr6r7、(ch2)mcho、(ch2)mco2r6、r6中的任何一种基团;r6和r7是相同或不同的基团,为h、cmh2m+1、cmh2m-1、cmh2m-3、c6+mh5+2m中的任一基团;m为1~20之间的整数。
一种耐酸性光控荧光分子开关,其优选的化学结构为:
其中:r1、r2、r3、r4、r5是相同或不同的取代基,具体为h、cmh2m+1、cmh2m-1、cmh2m-3、c6+mh5+2m、cmh2m+1co、cmh2m+1so2或cmh2m+1phso2中的任一基团;
x为o、c、si、ge、s或so2;
z为cmh2m+1、cmh2m-1、cmh2m-3、c6+mh5+2m、(c2h4o)mch3、(c2h4o)moh或cmh2m(c4h8no)中的任一基团;m是1~20之间的整数。
上述耐酸性光控荧光分子开关的合成方法具体步骤如下:
(1)将3-硝基罗丹明和烃基伯胺按物质的量比1:1-20溶解于无水乙醇,升温至回流,搅拌1-4小时后减压蒸除溶剂,中间体罗丹明3-硝基螺酰胺通过硅胶柱色谱分离提纯,
(2)取上述步骤(1)中的产物罗丹明3-硝基螺酰胺全部溶于体积比为1-5:1甲醇和二氯甲烷混合溶剂,在氢气氛围及占反应物质量百分比0.5-10%的钯碳催化下搅拌1-3小时,抽滤并取滤液,减压蒸除溶剂后得到罗丹明3-氨基螺酰胺产物;
(3)取上述步骤(2)中的产物罗丹明3-氨基螺酰胺和酰氯(或碘甲烷)按物质的量比1:1-30溶解于无水二氯甲烷,常温搅拌1-3小时后减压蒸除溶剂,最后将罗丹明3-酰胺取代的螺酰胺产物(或罗丹明3-单甲胺取代的螺酰胺产物)通过柱色谱分离提纯。
所述的烃基伯胺为丁胺、氨基六聚乙二醇、氨基六聚乙二醇单甲醚、或2-乙氨基吗啉;
所述的酰氯为乙酰氯、甲磺酰氯、或对甲苯磺酰氯。
其合成的路线为:
一种耐酸性光控荧光分子开关,其另外一个优选的化学结构式为:
其中:r1、r2、r3、r4、r5是相同或不同的取代基,为h、cmh2m+1、cmh2m-1、cmh2m-3、c6+mh5+2m、cmh2m+1co、cmh2m+1so2、cmh2m+1phso2中的任一基团;
x为o、c、si、ge、s或so2;
m为o、ncmh2m+1、nc6+mh5+2m、ncmh2m(c4h8no)、ncmh2m(c5h4n)、cmh2mco2nc4h4o2、c13h12n6o中的任一基团;m是1~20之间的整数。
上述耐酸性光控荧光分子开关的合成方法,该方法的具体步骤如下:
(1)将3-硝基罗丹明和三氯氧磷按物质的量比1:3-20溶解于1,2-二氯乙烷,升温至回流,搅拌1-3小时后蒸除溶剂,将粗酰氯中间体溶于无水二氯甲烷,随后逐滴加入三乙胺和6-(4-氨基苯乙炔基)萘酐混合溶液,其中粗酰氯中间体、三乙胺和6-(4-氨基苯乙炔基)萘酐三者的物质的量比为1:0.5-2:1-2,室温搅拌8-24小时后减压蒸除溶剂,中间体3-硝基取代罗丹明苯乙炔基萘酐螺酰胺经色谱柱提纯;
(2)取上述步骤(1)中的产物3-硝基取代罗丹明苯乙炔基萘酐螺酰胺与二水合氯化亚锡和浓盐酸分别按物质的量比1:1.5-5:0.1-2溶解于混合于无水乙醇中,回流搅拌5-8小时后减压蒸除溶剂,产物通过硅胶柱色谱分离提纯得到中间体3-氨基取代罗丹明苯乙炔基萘酐螺酰胺;
(3)取上述步骤(2)中的产物3-氨基取代罗丹明苯乙炔基萘酐螺酰胺与酰氯按物质的量比1:1-30混合于无水二氯甲烷中,室温搅拌反应0.5-3小时后通过硅胶柱色谱分离提纯,可得到3-酰胺取代罗丹明苯乙炔基萘酐螺酰胺;
(4)取上述步骤(3)中的产物将3-酰胺取代罗丹明苯乙炔基萘酐螺酰胺与伯胺分子按物质的量比例1:1-10混合并置于无水乙醇中回流2-10小时,最后通过硅胶柱色谱提纯即可得到3-酰胺取代罗丹明苯乙炔基萘酰亚胺螺酰胺。
所述的酰氯为乙酰氯、甲磺酰氯、或对甲苯磺酰氯;
所述的伯胺为丁胺、2-乙氨基吗啉、4-氨基甲基吡啶、4-氨甲基-1-(3-(2,5-二氧代吡咯烷基)氧代)-3-丙酰吡啶盐、或6-(4-氨甲基苄氧基)-9h-嘌呤-2-胺。
其合成的路线为:
一种耐酸性光控荧光分子开关的应用,基于耐酸性光控荧光分子开关的耐酸性和光控分子开关特点应用在超分辨荧光成像、生物及化学物质的传感及检测等诸多领域。
本发明的优点和有益效果为:
基于单分子定位的超分辨荧光成像技术的核心是荧光分子开关染料,而罗丹明螺酰胺是一类广泛应用于该技术的光空荧光分子开关染料。然而传统的罗丹明螺酰胺可以通过两种路径,即光控制或者酸控制,实现分子“开-关”的转变。然而生物环境普遍存在ph小于7的酸性环境,在酸性环境中这类染料发生酸控分子开关会导致其失去光控分子开关性能,因此这类染料在酸性环境中无法应用于的超分辨荧光成像技术。
而本发明里开发的3-伯胺或仲胺取代罗丹明螺酰胺不仅具有耐酸的性能,而且保留了光控分子开关性能(如图13所示)。因此这类耐酸性光控荧光分子开关可以应用于基于单分子定位的超分辨成像技术中,且不受生物环境中ph的干扰。此外,本发明中的耐酸光控荧光分子开关还可以作为分子荧光探针应用于传感及检测领域。
附图说明
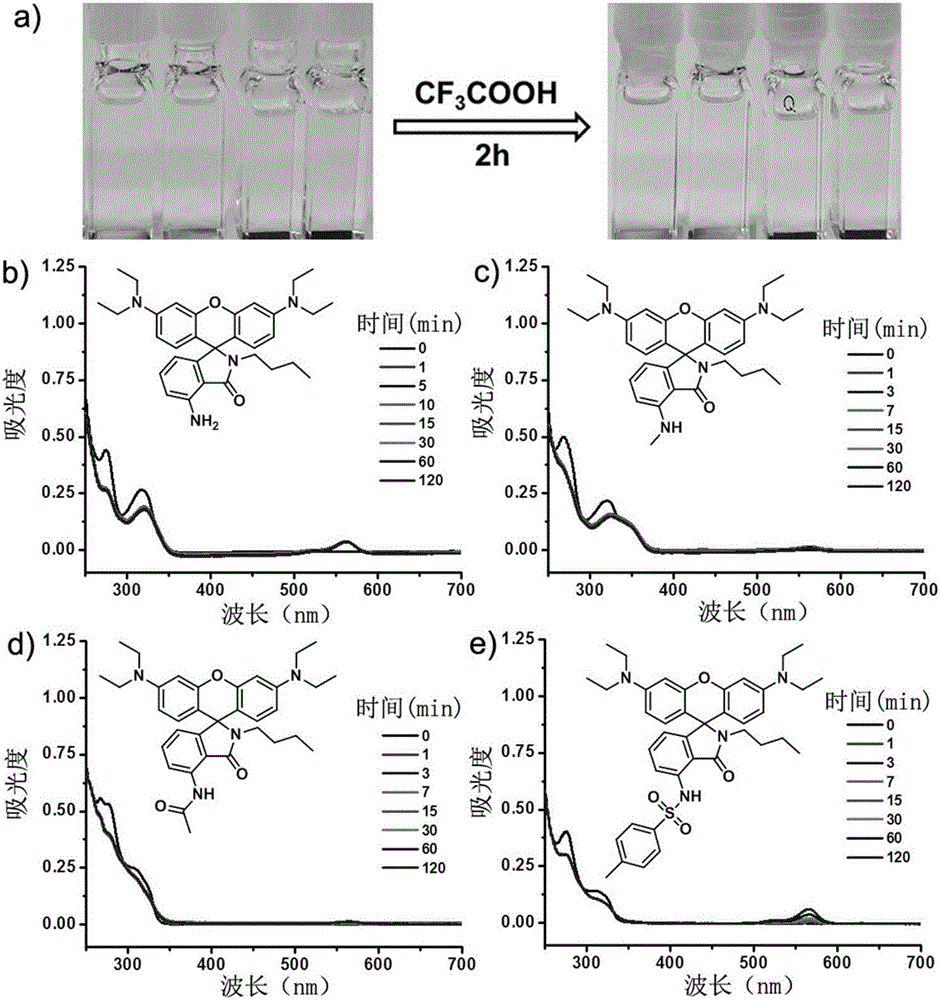
图1:为p1-p4在二氯甲烷/甲醇(9/1,v/v)混合溶剂中(浓度为10-5m)分别加入三氟乙酸(2.3μl,1000eq)前后的时间分辨紫外可见吸收光谱及可见照片变化;
图2:为水溶性产物(p6和p8)在不同ph值的缓冲溶液(浓度为10-5m)中测得的紫外可见吸收及荧光光谱;
图3:为p9(10μm)和商业的溶酶体标记染料(ltg,0.1μm)共同染色培养的mcf-7细胞置于不同紫外(375nm)光照时间下的共聚焦图像;
图4:为p10(10μm)和商业的溶酶体标记染料(ltg,0.1μm)共同染色培养的mcf-7细胞置于不同紫外(375nm)光照时间下的共聚焦图像;
图5:为p11-p14分别溶解在dmso溶液中测定的紫外可见吸收光谱,溶液浓度均为10-5m;
图6:p14的dmso溶液(浓度为10-5m)加入2.3μl三氟乙酸(1000eq)前后测得的时间分辨紫外可见吸收光谱;
图7:为掺杂有p14的聚乙烯醇薄膜,以405nm作为激活光(60w/cm2),测定分子随着激发光(561nm)功率密度增加相应的光激活性能参数的变化,包括总光子数(a),背景光子数(b)及定位精度(c);
图8:为掺杂有p14的聚乙烯醇薄膜,以405nm作为激活光(60w/cm2),在最优激发光561nm(1.2kw/cm2)下的光激活性能相关的参数包括总光子数(a),背景光子数(b)及定位精度(c),每帧图像中的光子数(d);
图9:p15标记的枯草芽孢杆菌表面的共聚焦成像及3d-storm超分辨荧光成像图。
图10:p16-p17分别溶解在dmso溶液中测定的紫外可见吸收光谱,溶液浓度均为10-5m;
图11:p16标记的u2os细胞中微管的宽场荧光图像及对应的2d-storm超分辨图像;
图12:p17标记的u2os细胞中微管的宽场荧光图像及对应的3d-storm超分辨图像;
图13:光诱导的耐酸性3-伯胺或仲胺取代的罗丹明螺酰胺分子的螺环及荧光开关的示意图。
具体实施方式
本发明给出了一种耐酸光控荧光分子开关的合成方法及其作为光激活荧光染料应用于基于单分子成像的超分辨荧光成像技术领域。
实施例1
当r1=r2=r3=r4=c2h5,r5=h,x=o,y=h,z=c4h9时,耐酸光控荧光分子开关p1合成路线和产物结构如下:
合成步骤及表征:将3-硝基罗丹明(5mmol,2.4g)和正丁胺(20mmol,1.4g)溶于无水乙醇(50ml)。升温至78℃回流,搅拌8小时后减压蒸除溶剂,产物通过硅胶色谱柱来分离提纯(石油醚/乙酸乙酯,8:1v/v),得到的浅黄色粉末(2.6g,95%)。接着将该粉末全部溶于甲醇/二氯甲烷(50ml,3:1v/v)混合溶剂中,在氢气氛围下,通过钯碳(0.21g,10%wt)催化还原,抽滤取滤液,减压蒸除溶剂后得到最终白色粉末产物(2g,98%)。
对白色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ7.14(t,j=7.6hz,1h),6.56(t,j=8.2hz,3h),6.41–6.25(m,5h),3.34(dd,j=13.4,6.5hz,8h),3.05(s,2h),1.24–1.04(m,16h),0.68(t,j=7.1hz,3h)。13cnmr(101mhz,cdcl3)δ169.59,154.86,153.08,148.63,144.95,133.24,129.03,114.15,113.31,112.11,108.04,106.77,97.69,,64.59,44.33,39.70,30.64,20.32,13.57,12.57ppm。lc-ms(esi):m/z:计算值:512.3151,实验值:513.3220[m+h]+。
经上述检测,鉴定其结构为p1所示。
产物p1溶解于二氯甲烷/甲醇(9/1,v/v)混合溶剂中(浓度为10-5m),往混合溶液中分别加入三氟乙酸(2.3μl,1000eq)。测试加酸前后时间分辨的紫外可见吸收光谱并拍摄可见光下的照片,如图1所示,加酸后p1没有出现罗丹明开环结构的特征吸收峰随酸化时间的延长而增强的现象,溶液颜色仍然保持无色,证明p1具有耐酸性。
实施例2
当r1=r2=r3=r4=c2h5,r5=ch3,x=o,y=h,z=c4h9时,其分子(p2)合成路线和产物结构如下:
合成步骤及表征:将p1(0.25g,0.5mmol),碘甲烷(0.28g,2mmol)和碳酸钾(0.34g,2.5mmol)混合于乙腈(8ml),回流搅拌10小时,冷却至室温后过滤得到滤液,减压蒸除溶剂,粗产物通过柱色谱(硅胶,石油醚/乙酸乙酯,10:1v/v)分离提纯得到白色粉末p2(0.17g,65%)。
对白色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ7.23(t,j=7.9hz,1h),6.75(d,j=4.9hz,1h),6.57(t,j=9.3hz,2h),6.49(d,j=8.1hz,1h),6.41–6.22(m,5h),3.33(q,j=7.0hz,8h),3.04(s,2h),2.97(d,j=4.9hz,3h),1.16(t,j=6.9hz,12h),1.07(s,4h),0.67(t,j=6.5hz,3h)。13cnmr(101mhz,cdcl3)δ170.15,154.93,153.09,148.60,147.23,133.78,129.01,113.08,110.19,108.01,107.53,106.74,97.67,44.33,39.63,30.69,29.42,20.30,13.60,12.57。lc-ms(esi):m/z:计算值:526.3308;实验值:527.3523[m+h]+。
经上述检测,鉴定其结构为p2所示。
产物p2溶解于二氯甲烷/甲醇(9/1,v/v)混合溶剂中(浓度为10-5m),往混合溶液中分别加入三氟乙酸(2.3μl,1000eq)。测试加酸前后时间分辨的紫外可见吸收光谱并拍摄可见光下的照片,如图1所示,加酸后p2没有出现罗丹明开环结构的特征吸收峰随酸化时间的延长而增强的现象,溶液颜色仍然保持无色,证明p2具有耐酸性。
实施例3
当r1=r2=r3=r4=c2h5,r5=ch3co,x=o,y=h,z=c4h9时,其分子(p3)合成路线和产物结构如下:
合成步骤及表征:将p1(0.25g,0.5mmol)和乙酰氯(58mg,0.75mmol)混合于二氯甲烷(5ml),搅拌2小时后减压蒸除溶剂,粗产物通过柱色谱(硅胶,石油醚/乙酸乙酯,8:1v/v)分离提纯得到白色粉末p3(0.26g,95%)。
对白色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ10.60(s,1h),8.43(d,j=8.2hz,1h),7.39(t,j=7.9hz,1h),6.74(d,j=7.6hz,1h),6.46(d,j=8.8hz,2h),6.38(d,j=2.6hz,2h),6.28(dd,j=8.9,2.6hz,2h),3.34(q,j=7.0hz,8h),3.06(t,j=7.0hz,2h),2.29(s,3h),1.17(t,j=7.0hz,12h),1.12–1.02(m,4h),0.69(t,j=6.7hz,3h)。13cnmr(101mhz,cdcl3)δ169.30,168.85,158.27,153.50,153.27,148.83,136.75,133.81,128.78,117.95,117.52,116.43,108.07,105.32,101.26,99.97,97.72,65.17,44.36,39.98,30.41,24.97,20.35,13.55,12.55。lc-ms(esi):m/z:计算值:554.3257;实验值:555.3382[m+h]+。
经上述检测,鉴定其结构为p3所示。
产物p3溶解于二氯甲烷/甲醇(9/1,v/v)混合溶剂中(浓度为10-5m),往混合溶液中分别加入三氟乙酸(2.3μl,1000eq)。测试加酸前后时间分辨的紫外可见吸收光谱并拍摄可见光下的照片,如图1所示,加酸后p3没有出现罗丹明开环结构的特征吸收峰随酸化时间的延长而增强的现象,证明p3具有耐酸性。
实施例4
当r1=r2=r3=r4=c2h5,r5=ch3phso2,x=o,y=h,z=c4h9时,其分子(p4)合成路线和产物结构如下:
合成步骤及表征:将p1(0.25g,0.5mmol)和对甲苯磺酰氯(95mg,0.5mmol)混合于二氯甲烷(5ml),搅拌3小时后减压蒸除溶剂,粗产物通过柱色谱(硅胶,石油醚/乙酸乙酯,6:1v/v)分离得到黄色粉末p4(0.30g,91%)。
对黄色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ9.88(s,1h),7.83(d,j=8.3hz,2h),7.51(d,j=8.1hz,1h),7.32–7.25(m,3h),6.67(d,j=7.5hz,1h),6.35(d,j=1.9hz,2h),6.28–6.14(m,4h),3.33(q,j=7.1hz,8h),2.99(t,j=7.0hz,2h),2.41(s,3h),1.16(t,j=7.0hz,12h),1.07–0.96(m,4h),0.67(t,j=6.8hz,3h)。13cnmr(101mhz,cdcl3)δ167.92,153.86,153.14,148.78,143.49,136.52,135.47,133.45,129.43,128.58,127.52,118.59,118.03,117.21,107.90,105.02,97.67,65.03,44.34,39.76,30.25,21.55,20.19,13.54,12.50。lc-ms(esi):m/z:计算值:666.3240;实验值:667.3211[m+h]+。
经上述检测,鉴定其结构为p4所示。
产物p4溶解于二氯甲烷/甲醇(9/1,v/v)混合溶剂中(浓度为10-5m),往混合溶液中分别加入三氟乙酸(2.3μl,1000eq)。测试加酸前后时间分辨的紫外可见吸收光谱并拍摄可见光下的照片,如图1所示,加酸后p4没有出现罗丹明开环结构的特征吸收峰随酸化时间的延长而增强的现象,证明p4具有耐酸性。
实施例5
当r1=r2=r3=r4=c2h5,r5=h,x=o,y=h,z=c6h5时,其分子(p5)合成路线和产物结构如下:
合成步骤及表征:将3-硝基罗丹明(2mmol,1.12g)和苯胺(2mmol,0.186g)溶于无水乙醇(5ml)。升温至78℃回流,搅拌4小时后减压蒸除溶剂,产物通过柱色谱(硅胶,石油醚/乙酸乙酯,6:1v/v)分离得到的浅黄色固体(1.08g,96%)。接着将该固体产物全部溶于甲醇和二氯甲烷(5ml,3:1v/v)混合溶剂中,在氢气氛围及钯碳(10%wt)催化下搅拌1小时,抽滤并取滤液,减压蒸除溶剂后得到最终白色固体产物(1.01g,99%)。
对白色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ7.22(t,j=7.7hz,1h),7.14-7.04(m,3h),6.85-7.74(m,4h),6.60(d,j=7.9hz,1h),6.40(d,j=7.4hz,1h),6.33(dd,j=8.8,2.5hz,2h),6.24(d,j=2.5hz,2h),5.41(s,2h),3.31(q,j=7.1hz,8h),1.14(t,j=7.0hz,12h)。13cnmr(101mhz,cdcl3)δ169.30,154.56,152.84,148.60,145.57,136.55,133.97,128.86,128.40,127.15,126.32,113.40,113.33,112.15,108.03,107.08,97.67,67.10,44.25,12.55。lc-ms(esi):m/z:计算值:532.2838,实验值:533.2840[m+h]+。
经上述检测,鉴定其结构为p5所示。
产物p5溶解于二氯甲烷/甲醇(9/1,v/v)混合溶剂中(浓度为10-5m),往混合溶液中分别加入三氟乙酸(2.3μl,1000eq)。测试加酸前后时间分辨的紫外可见吸收光谱并拍摄可见光下的照片,结果显示加酸后p5没有出现罗丹明开环结构的特征吸收峰,溶液颜色仍然保持无色,证明p5具有耐酸性。
实施例6
当r1=r2=r3=r4=c2h5,r5=h,x=o,y=h,z=peg6-oh(c12h25o6)时,其分子(p6)合成路线和产物结构如下:
合成步骤及表征:将3-硝基罗丹明(2mmol,0.974g)和氨基六聚乙二醇(2mmol,0.562g)溶于无水乙醇(5ml),升温至78℃回流,搅拌4小时后减压蒸除溶剂,产物通过柱色谱(硅胶,二氯甲烷/甲醇,10:1v/v)分离提纯得到的浅黄色粘性液体(1.42g,95%)。接着将该液体产物全部溶于甲醇(5ml),在氢气氛围及钯碳(10%wt)催化下搅拌1小时,抽滤并取滤液,减压蒸除溶剂后得到最终白色粘性液体产物(1.35g,99%)。
对粘性液体产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ7.14(t,j=7.7hz,1h),6.58–6.50(m,3h),6.38–6.26(m,5h),3.74–3.70(m,2h),3.67–3.56(dd,j=16.6,8.3hz,16h),3.51–3.47(m,2h),3.39–3.26(m,12h),3.17–3.09(m,4h),1.16(t,j=7.0hz,12h)。13cnmr(101mhz,cdcl3)δ169.84,155.00,152.99,148.64,145.12,133.59,128.88,113.41,112.01,108.03,106.16,97.98,97.65,72.62,72.60,70.45,70.38,70.37,70.28,70.25,70.13,69.93,69.90,68.07,64.55,61.50,44.33,12.61。lc-ms(esi):m/z:计算值:720.4098,实验值:721.4183[m+h]+。
经上述检测,鉴定其结构为p6所示。
将水溶性产物p6溶解于不同ph值的缓冲溶液(浓度为10-5m)中,并测试其在不同ph下的紫外可见吸收光谱和荧光光谱(图2)。如图2所示,p6在ph为酸性的缓冲溶液中并没有出现罗丹明的特征吸收峰和发射峰,这表明在酸性环境中闭环螺酰胺结构没有发生改变,进一步证明了p6的耐酸性特征。
实施例7
当r1=r2=r3=r4=c2h5,r5=h,x=o,y=h,z=peg6-ch3(c13h27o6)时,其分子(p7)合成路线和产物结构如下:
合成步骤及表征:3-硝基罗丹明(0.24g,0.5mmol)和氨基六聚乙二醇单甲醚(0.14g,0.5mmol)溶于无水乙醇(8ml)中,升温至78℃回流,搅拌4小时后减压蒸除溶剂,残余物通过柱色谱(硅胶,乙酸乙酯/甲醇,30:1v/v)分离得到粘性液体(0.34g,90%)。将其全部溶于甲醇(5ml)并加入20mg10%钯碳在氢气氛围下催化还原。反应混合物抽滤,滤液通过减压蒸除溶剂,产物通过柱色谱(硅胶,乙酸乙酯/甲醇,20:1v/v)分离得到粘性液体p7(0.32g,96%)。
对粘性液体产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ7.11(t,j=7.7hz,1h),6.54(t,j=8.9hz,3h),6.35(d,j=2.4hz,2h),6.32–6.23(m,3h),5.37(s,2h),3.67–3.51(m,16h),3.51–3.46(m,2h),3.41–3.25(m,15h),3.13(t,j=7.2hz,2h),1.15(t,j=7.0hz,12h)。13cnmr(101mhz,cdcl3)δ169.55,154.76,152.76,148.38,144.91,133.28,128.65,113.20,113.08,111.69,107.79,106.04,97.44,71.66,70.32,70.27,70.24,70.24,70.22,70.11,69.70,67.85,64.26,58.74,44.09,38.56,12.38。lc-ms(esi):m/z:计算值:734.4255;实验值:735.4290[m+h]+。
经上述检测,鉴定其结构为p7所示。
将水溶性产物p7溶解于不同ph值的缓冲溶液(浓度为10-5m)中,并测试其在不同ph下的紫外可见吸收光谱和荧光光谱。结果显示p7在ph为酸性的缓冲溶液中并没有出现罗丹明的特征吸收峰和发射峰,这表明在酸性环境中闭环螺酰胺结构没有发生改变,进一步证明了p7的耐酸性特征。
实施例8
当r1=r2=r3=r4=c2h5,r5=ch3co,x=o,y=h,z=peg6-ch3(c13h27o6)时,其分子(p8)合成路线和产物结构如下:
合成步骤及表征:将p7(0.22g,0.3mmol)和乙酰氯(35mg,0.45mmol)混合于二氯甲烷(5ml)中,搅拌2小时后减压蒸除溶剂并通过柱色谱(硅胶,乙酸乙酯/甲醇,20:1v/v)分离得到粘性液体p8(0.22g,95%)。
对粘性液体产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ10.47(s,1h),8.39(d,j=8.2hz,1h),7.34(t,j=7.9hz,1h),6.68(d,j=7.6hz,1h),6.41(d,j=8.8hz,2h),6.33(d,j=2.4hz,2h),6.24(dd,j=8.9,2.4hz,2h),3.60–3.48(m,16h),3.47–3.42(m,2h),3.35–3.25(m,15h),3.08(t,j=7.1hz,2h),2.25(s,3h),1.13(t,j=7.0hz,13h)。13cnmr(101mhz,cdcl3)δ169.06,168.82,153.52,152.97,148.66,136.57,133.85,128.43,117.79,117.38,115.71,107.90,104.58,97.56,71.69,70.34,70.30,70.11,69.82,67.62,64.95,58.78,44.15,38.83,24.73,12.38。lc-ms(esi):m/z:计算值:776.4360;实验值:777.4435[m+h]+。
经上述检测,鉴定其结构为p8所示。
将水溶性产物p8溶解于不同ph值的缓冲溶液(浓度为10-5m)中,并测试其在不同ph下的紫外可见吸收光谱和荧光光谱。如图2所示,p8在ph为酸性的缓冲溶液中并没有出现罗丹明的特征吸收峰和发射峰,这表明在酸性环境中闭环螺酰胺结构没有发生改变,进一步证明了p8的耐酸性特征。
实施例9
当r1=r2=r3=r4=c2h5,r5=h,x=o,y=h,z=c6h12no时,其分子(p9)合成路线和产物结构如下:
合成步骤及表征:将3-硝基罗丹明(2mmol,0.974g)和2-乙氨基吗啉(2mmol,0.146g)溶于无水乙醇(35ml)。升温至78℃回流,搅拌4小时后减压蒸除溶剂,产物通过柱色谱(硅胶,石油醚/乙酸乙酯,4:1v/v),分离提纯最后得到的浅黄色粉末(1.14g,95%)。接着将该粉末全部溶于甲醇(5ml),在氢气氛围及钯碳(10%wt)催化下搅拌1小时,抽滤取滤液,减压蒸除溶剂后得到最终白色粉末产物p9(1.07g,99%)。
对白色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ7.15(t,j=7.7hz,1h),6.56(dd,j=8.3,5.8hz,3h),6.34(t,j=5.3hz,3h),6.28(dd,j=8.9,2.6hz,2h),3.63–3.52(m,4h),3.33(q,j=7.0hz,8h),3.24–3.15(m,2h),2.24(s,4h),2.11–2.04(m,2h),1.16(t,j=7.0hz,12h).13cnmr(101mhz,cdcl3)δ169.45,154.71,153.07,148.58,144.93,133.41,129.08,113.91,113.36,112.15,107.97,106.32,97.55,66.89,64.52,56.33,53.22,44.32,36.55,12.54.lc-ms(esi):m/z:计算值:569.3366,实验值:570.3457[m+h]+。
经上述检测,鉴定其结构为p9所示。
将p9溶解于二氯甲烷/甲醇(9/1,v/v)混合溶剂中(浓度为10-5m),往溶液中加入三氟乙酸(2.3μl,1000eq)。测试加酸前后时间分辨的紫外可见吸收光谱并拍摄可见光下的照片,结果所示加酸后p9没有出现罗丹明开环结构的特征吸收峰随酸化时间的延长而增强的现象,溶液颜色仍然保持无色,证明p9具有耐酸性。
将p9(10μm)和商业的溶酶体标记染料(ltg,0.1μm)共同染色培养mcf-7细胞,通过激光共聚焦倒置显微镜实时观察两个通道内的荧光染色状况,绿色通道的激发光波长为488nm,采集500–550nm波段的荧光信号,红色通道的激发光波长561nm,采集580–653nm波段的荧光信号。对比观察发现绿色通道在染色0.5小时后就能够观察到溶酶体中的荧光信号,而红色通道在染色2小时后溶酶体中仍然没有出现明显的荧光信号,随后用375nm紫外光原位辐照细胞,分别采集辐照0和3分钟各自两个通道的荧光图像(图3),对比发现随着紫外辐照时间的延长,红色通道中溶酶体内的荧光信号由弱变强,与绿色通道中的荧光信号能够很好重叠,这些结果表明p9染料在生物酸性环境中能够保持耐酸的特性,同时在酸性环境中具有光激活荧光的性能。
实施例10
当r1=r2=r3=r4=c2h5,r5=ch3co,x=o,y=h,z=c6h12no时,其分子(p10)合成路线和产物结构如下:
合成步骤及表征:将p9(1.14g,2mmol)和乙酰氯(0.23g,3mmol)混合于二氯甲烷(30ml)中,搅拌2小时后减压蒸除溶剂,粗产物通过柱色谱(硅胶,石油醚/乙酸乙酯,8:1v/v)提纯得到白色粉末p10(1.16g,95%)。
对白色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ10.52(s,1h),8.45(d,j=8.2hz,1h),7.40(t,j=7.9hz,1h),6.75(d,j=7.6hz,1h),6.47(d,j=8.8hz,2h),6.37(d,j=2.5hz,2h),6.28(dd,j=8.9,2.5hz,2h),3.62–3.51(m,4h),3.34(q,j=7.0hz,8h),3.25–3.16(m,2h),2.29(s,3h),2.23(s,4h),2.10–2.03(m,2h),1.16(t,j=7.0hz,12h).13cnmr(101mhz,cdcl3)δ169.30,168.76,153.38,153.30,148.85,136.77,133.97,128.87,118.03,117.62,116.29,108.08,105.01,97.67,66.87,65.14,56.07,53.22,44.37,36.82,29.67,24.97,12.53.lc-ms(esi):m/z:计算值:611.3472,实验值:612.3507[m+h]+。
经上述检测,鉴定其结构为p10所示。
将p10溶解于二氯甲烷/甲醇(9/1,v/v)混合溶剂中(浓度为10-5m),往溶液中加入三氟乙酸(2.3μl,1000eq)。测试加酸前后时间分辨的紫外可见吸收光谱并拍摄可见光下的照片,结果所示加酸后p10没有出现罗丹明开环结构的特征吸收峰随酸化时间的延长而增强的现象,溶液颜色仍然保持无色,证明p10具有耐酸性。
用p10(10μm)和商业的溶酶体标记染料(ltg,0.1μm)共同染色培养mcf-7细胞,通过激光共聚焦倒置显微镜实时观察两个通道内的荧光染色状况,绿色通道的激发光波长为488nm,采集500–550nm波段的荧光信号,红色通道的激发光波长561nm,采集580–653nm波段的荧光信号。对比观察发现绿色通道在染色0.5小时后就能够观察到溶酶体中的荧光信号,而红色通道在染色2小时后溶酶体中仍然没有出现明显的荧光信号,随后用375nm紫外光原位辐照细胞,分别采集辐照0,1和5分钟各自两个通道的荧光图像(图4),对比发现随着紫外辐照时间的延长,红色通道中溶酶体内的荧光信号从无到有并逐渐增强,与绿色通道中的荧光信号能够很好重叠,这些结果表明p10染料在生物酸性环境中能够保持耐酸的特性,同时在酸性环境中具有光激活荧光的性能。
实施例11
当r1=r2=r3=r4=c2h5,r5=h,x=o,y=h,z=6-(4-苯乙炔基)萘酐(c20h10o3)时,其分子(p11)合成路线和产物结构如下:
合成步骤及表征:将3-硝基罗丹明(2.92g,6mmol)和三氯氧磷(5.6ml,60mmol)置于1,2-二氯乙烷(150ml),升温至84℃回流,搅拌2小时后蒸除溶剂得到暗紫红色油状液体。将粗酰氯产物溶于二氯甲烷(100ml),随后逐滴加入三乙胺(3ml)和6-(4-氨基苯乙炔基)萘酐(1.88g,6mmol)混合溶液中,室温搅拌24小时后减压蒸除溶剂,残余物通过柱色谱(硅胶,二氯甲烷/乙酸乙酯,30:1v/v)分离得到黄色粉末状中间体(2.44g,52%)。取上述黄色粉末(1.56g,2mmol),二水合氯化亚锡(1.80g,8mmol)和浓盐酸(9ml)置于无水乙醇(50ml)中升温至78℃回流,搅拌8小时后减压蒸除溶剂,粗产物通过柱色谱(硅胶,乙酸乙酯/石油醚,1:3v/v)分离得到黄色固体p11(1.27g,85%)。
对黄色固体产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ8.75(d,j=8.3hz,1h),8.64(d,j=7.2hz,1h),8.54(d,j=7.7hz,1h),7.90(d,j=7.7hz,1h),7.85(t,j=7.8hz,1h),7.44(d,j=8.5hz,2h),7.22(t,j=7.7hz,1h),7.13(d,j=8.6hz,2h),6.76(d,j=8.5hz,2h),6.60(d,j=8.0hz,1h),6.37(d,j=7.4hz,1h),6.35–6.24(m,4h),5.44(s,2h),3.32(q,j=7.0hz,8h),1.16(t,j=7.0hz,12h)。13cnmr(101mhz,cdcl3)δ169.69,160.45,160.16,154.88,152.57,148.73,145.74,138.85,134.52,133.91,133.73,132.58,132.28,131.66,130.76,130.19,129.48,128.53,127.77,125.45,119.00,118.41,117.57,113.36,112.35,111.95,108.16,106.83,101.06,97.69,85.74,67.17,44.26,12.57。lc-ms(esi):m/z:计算值:752.2999;实验值:753.3073[m+h]+。
经上述检测,鉴定其结构为p11所示。
测试产物p11的dmso溶液(浓度为10-5m)的紫外可见吸收光谱,如图5所示p11最大吸收波长约为400nm。
向p11的dmso溶液(浓度为10-5m)中加入2.3μl三氟乙酸(1000eq),测定加酸前后时间分辨的紫外可见吸收光谱,结果显示p11的最大吸收波长处的吸光度没有随着酸化时间的增长而增加,表明p11分子仍然具有耐酸的特性。
将产物p11以单分子形式掺杂到聚乙烯醇水溶液(p11浓度约为10nm),经涂膜固化后制成50nm的膜层材料,随后以405nm作为激活光(60w/cm2),测定薄膜中p11分子随着激发光(561nm)功率密度增加相应的光激活性能参数的变化,包括总光子数,背景光子数及定位精度,结果显示p11具有可激光激活性能。
实施例12
当r1=r2=r3=r4=c2h5,r5=ch3co,x=o,y=h,m=6-(4-苯乙炔基)萘酐(c20h10o3)时,其分子(p12)合成路线和产物结构如下:
合成步骤及表征:将p11(0.75g,1mmol)和乙酰氯(0.12g,1.5mmol)混合于二氯甲烷(10ml),搅拌2小时后减压蒸除溶剂,粗产物通过柱色谱(硅胶,乙酸乙酯/石油醚,1:3v/v)分离得到黄色粉末产物p12(0.76g,96%)。
对黄色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ10.58(s,1h),8.75(d,j=8.2hz,1h),8.65(d,j=7.2hz,1h),8.55(d,j=7.7hz,1h),8.51(d,j=8.2hz,1h),7.92(d,j=7.7hz,1h),7.90–7.82(m,1h),7.56–7.43(m,3h),7.00(d,j=8.5hz,2h),6.81(d,j=7.6hz,1h),6.67(d,j=8.8hz,2h),6.37–6.26(m,4h),3.33(q,j=7.0hz,8h),2.31(s,3h),1.17(t,j=7.0hz,12h)。13cnmr(100mhz,cdcl3)δ169.31,168.94,160.38,160.11,153.44,152.94,148.99,137.79,137.43,134.99,133.82,133.77,132.55,132.43,131.70,130.91,130.20,129.24,128.48,127.84,126.34,119.68,119.10,118.08,117.81,115.18,108.26,105.41,100.48,97.77,86.08,67.99,44.32,24.98,12.55。lc-ms(esi):m/z:计算值:794.3104;实验值:795.3177[m+h]+。
经上述检测,鉴定其结构为p12所示。
测试产物p12的dmso溶液(浓度为10-5m)的紫外可见吸收光谱,如图5所示p12最大吸收波长约为400nm。
向p12的dmso溶液(浓度为10-5m)中加入2.3μl三氟乙酸(1000eq),测定加酸前后时间分辨的紫外可见吸收光谱,结果显示p12的最大吸收波长处的吸光度没有随着酸化时间的增长而增加,表明p12分子仍然具有耐酸的特性。
将产物p12以单分子形式掺杂到聚乙烯醇水溶液(p12浓度约为10nm),经涂膜固化后制成50nm的膜层材料,随后以405nm作为激活光(60w/cm2),测定薄膜中p12分子随着激发光(561nm)功率密度增加相应的光激活性能参数的变化,包括总光子数,背景光子数及定位精度,结果显示p12具有可激光激活性能。
实施例13
当r1=r2=r3=r4=c2h5,r5=h,x=o,y=h,m=1-(2-乙氨基吗啉)-6-(4-苯乙炔基)萘酰亚胺(c26h22n2o3)时,其分子(p13)合成路线和产物结构如下:
合成步骤及表征:将p11(0.37g,0.5mmol)和2-乙氨基吗啉(0.19g,1.5mmol)混合于无水乙醇(10ml)中,升温至78℃回流,搅拌10小时后减压蒸除溶剂,残余物通过柱色谱(硅胶,乙酸乙酯/石油醚,1:2v/v)分离得到黄色粉末产物p13(0.36g,84%)。
对黄色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ8.66(d,j=8.3hz,1h),8.61(d,j=7.1hz,1h),8.52(d,j=7.7hz,1h),7.87(d,j=7.6hz,1h),7.79(t,j=7.8hz,1h),7.44(d,j=8.4hz,2h),7.22(t,j=7.7hz,1h),7.10(d,j=8.5hz,2h),6.76(d,j=8.6hz,2h),6.60(d,j=8.0hz,1h),6.37(d,j=7.4hz,1h),6.32(d,j=10.4hz,4h),5.44(s,2h),4.34(t,j=6.8hz,2h),3.68(s,4h),3.32(dd,j=13.9,6.8hz,7h),2.70(t,j=6.7hz,2h),2.60(s,4h),1.16(t,j=6.9hz,12h)。13cnmr(101mhz,cdcl3)δ169.69,164.07,163.79,160.61,157.75,154.91,152.65,148.78,145.75,138.50,134.50,132.53,132.22,131.62,131.60,130.55,130.46,128.62,128.13,127.89,127.39,125.65,122.86,121.79,119.00,113.39,112.54,112.04,108.20,106.90,97.75,86.27,67.22,67.04,56.15,53.82,44.32,37.25,12.63。lc-ms(esi):m/z:计算值:864.3999,实验值:865.4061[m+h]+。
经上述检测,鉴定其结构为p13所示。
测试产物p13的dmso溶液(浓度为10-5m)的紫外可见吸收光谱,如图5所示p13最大吸收波长约为400nm。
向p13的dmso溶液(浓度为10-5m)中加入2.3μl三氟乙酸(1000eq),测定加酸前后时间分辨的紫外可见吸收光谱,结果显示p13的最大吸收波长处的吸光度没有随着酸化时间的增长而增加,表明p13分子仍然具有耐酸的特性。
将产物p13以单分子形式掺杂到聚乙烯醇水溶液(p13浓度约为10nm),经涂膜固化后制成50nm的膜层材料,随后以405nm作为激活光(60w/cm2),测定薄膜中p13分子随着激发光(561nm)功率密度增加相应的光激活性能参数的变化,包括总光子数,背景光子数及定位精度,结果显示p13具有可激光激活性能。
实施例14
当r1=r2=r3=r4=c2h5,r5=ch3co,x=o,y=h,m=1-(4-亚甲基吡啶)-6-(4-苯乙炔基)萘酰亚胺(c26h16n2o2)时,其分子(p14)合成路线和产物结构如下:
合成步骤及表征:p12(0.40g,0.5mmol)和4-氨基甲基吡啶(0.15ml,1.5mmol)混合于无水乙醇(10ml),升温至78℃回流,搅拌8小时后减压蒸除溶剂,残余物通过柱色谱(硅胶,二氯甲烷/甲醇,20:1v/v)分离提纯得到黄色粉末产物p14(0.42g,96%)。
对黄色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,cdcl3)δ10.59(s,1h),8.66(dd,j=16.5,7.7hz,2h),8.52(d,j=9.9hz,4h),7.89(d,j=7.5hz,1h),7.81(t,j=7.7hz,1h),7.48(dd,j=13.1,8.0hz,3h),7.37(d,j=4.3hz,2h),6.98(d,j=8.1hz,2h),6.81(d,j=7.4hz,1h),6.67(d,j=8.7hz,2h),6.42–6.23(m,4h),5.36(s,2h),3.33(q,j=7.0hz,8h),2.30(s,3h),1.16(t,j=6.6hz,12h)。13cnmr(101mhz,cdcl3)δ169.29,168.89,163.89,163.61,153.41,152.95,149.99,148.97,145.77,137.50,137.41,134.93,132.83,132.34,132.04,131.60,130.83,130.67,128.48,128.12,127.48,126.40,123.21,122.49,121.48,120.04,118.08,117.78,115.25,108.23,105.40,99.36,97.76,86.49,67.99,44.30,42.64,24.97,12.54。lc-ms(esi):m/z:计算值:884.3686;实验值:885.3804[m+h]+。
经上述检测,鉴定其结构为p14所示。
测试产物p14的dmso溶液(浓度为10-5m)的紫外可见吸收光谱,如图5所示p14最大吸收波长约为400nm。
向p14的dmso溶液(浓度为10-5m)中加入2.3μl三氟乙酸(1000eq),测定加酸前后时间分辨的紫外可见吸收光谱(图6),如图6所示p14的最大吸收波长处的吸光度没有随着酸化时间的增长而增加,表明p14分子仍然具有耐酸的特性。
将产物p14以单分子形式掺杂到聚乙烯醇水溶液(p14浓度约为10nm),经涂膜固化后制成50nm的膜层材料,随后以405nm作为激活光(60w/cm2),测定薄膜中p14分子随着激发光(561nm)功率密度增加相应的光激活性能参数的变化(图7),包括总光子数(7a),背景光子数(7b)及定位精度(7c);并给出以405nm作为激活光(60w/cm2),在最优激发光561nm(1.2kw/cm2)下的光激活性能相关的参数(图8),包括总光子数(8a),背景光子数(8b)及定位精度(8c),每帧图像中的光子数(8d)。上述结果显示p14具有可激光激活性能。
实施例15
当r1=r2=r3=r4=c2h5,r5=ch3co,x=o,y=h,m=(c33h24n3o6)+i-时,其分子(p15)合成路线和产物结构如下:
合成步骤及表征:p14(0.26g,0.3mmol)和3-碘丙酸琥珀酰亚胺酯(0.1g,0.35mmol)混合于无水乙腈(10ml),升温至82℃回流,搅拌24小时后减压蒸除溶剂,残余物置于乙酸乙酯中搅拌过夜,抽滤得到棕色粉末产物p15(0.26g,74%)。
对棕色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,dmso)δ10.41(s,1h),9.02(d,j=5.8hz,2h),8.83(d,j=8.0hz,1h),8.58(d,j=7.0hz,1h),8.49(d,j=7.5hz,1h),8.34(d,j=8.0hz,1h),8.24(d,j=5.8hz,2h),8.07(d,j=7.4hz,1h),8.04–7.94(m,1h),7.66(d,j=8.1hz,2h),7.60–7.49(m,1h),7.09(d,j=7.6hz,2h),6.73(d,j=6.8hz,2h),6.62–6.12(m,4h),5.50(s,2h),4.92(t,2h),3.63(t,2h),3.35(q,j=7.0hz,8h),2.80(s,4h),2.25(s,3h),1.08(t,j=6.1hz,12h)。13cnmr(101mhz,dmso)δ169.99,168.86,168.05,166.49,163.52,163.23,157.90,151.96,145.00,137.69,137.05,135.13,132.44,132.38,131.53,130.98,130.93,130.25,128.36,127.94,126.44,125.39,122.76,122.12,119.02,117.65,117.38,114.52,113.45,98.45,86.69,66.59,54.76,42.98,31.27,25.45,24.63,12.17。lc-ms(esi):m/z:计算值:1054.4134;实验值:1054.4212[m]+。
经上述检测,鉴定其结构为p15所示。
测试产物p15的dmso溶液(浓度为10-5m)的紫外可见吸收光谱,结果显示p5最大吸收波长约为400nm。
向p15的dmso溶液(浓度为10-5m)中加入2.3μl三氟乙酸(1000eq),测定加酸前后时间分辨的紫外可见吸收光谱,结果显示p15的最大吸收波长处的吸光度没有随着酸化时间的增长而增加,表明p15分子仍然具有耐酸的特性。
将野生枯草芽孢杆菌的菌株放置于5mllb培养基中37℃摇床摇动培养过夜,取1ml细菌培养液至于1.5ml离心管中离心,转速10000转/分,离心时间3min,离心结束后弃去上层清液,往离心管中里加入1mlpbs(ph=4.5)缓冲溶液再悬浮后接着离心并去掉上层清液。接着将p15配成浓度为10-8m的dmso溶液,取50ul染料的dmso母液稀释到950ulpbs(ph=4.5)缓冲溶液中,取上述混匀的母液1ml加入细菌离心管中再悬浮后置于37℃摇床摇动培养30min,随后重复上述离心-再悬浮过程7-10次,最后取20ul细菌培养液滴到琼脂糖凝胶上,将接种有细菌的琼脂糖盖到用氩等离子体清洗过的载玻片上,即可置于超分辨storm显微镜下成像。选择常用的405nm激光作为激活光,561nm激光作为激发光,通过随机光激活以及重叠重构技术即可得到超分辨图像。图9为p15标记的枯草芽孢杆菌表面后分别在激光共聚焦显微镜和超分辨storm显微镜下拍摄的图像。
实施例16
当r1=r2=r3=r4=c2h5,r5=h,x=o,y=h,z=1-(4-苄基鸟嘌呤)-6-(4-苯乙炔基)萘酰亚胺(c33h22n6o3)时,其分子(p16)合成路线和产物结构如下:
合成步骤及表征:p11(0.37g,0.5mmol)和bg-nh2(0.19g,1.5mmol)混合于无水乙醇(10ml),升温至78℃回流,搅拌10小时后减压蒸除溶剂。粗产物通过柱色谱(硅胶,乙酸乙酯/石油醚,1:2v/v)分离提纯得到黄色粉末产物p16(0.42g,84%)。
对黄色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,dmso)δ12.53(s,1h),8.75(d,j=8.4hz,1h),8.56(d,j=7.3hz,1h),8.46(d,j=7.7hz,1h),8.01(d,j=7.7hz,1h),7.99–7.90(m,1h),7.76(s,1h),7.58(d,j=8.7hz,2h),7.40(dd,j=21.1,8.1hz,3h),7.32–7.09(m,3h),6.63(dd,j=10.1,6.4hz,2h),6.46(s,2h),6.37(dd,j=8.9,2.4hz,1h),6.32(d,j=2.4hz,1h),6.26(s,2h),6.13–5.96(m,2h),5.68(dd,j=8.9,2.9hz,1h),5.41(s,2h),5.25(s,2h),3.29(dd,j=14.2,7.0hz,8h),2.54(s,2h),1.06(t,j=6.9hz,12h)。lc-ms(esi):m/z:计算值:1004.4122,实验值:503.2012[m+2h]2+/2。
经上述检测,鉴定其结构为p16所示。
将产物p16溶于dmso中配成溶液(浓度为10-5m),测试溶液的紫外可见吸收光谱,如图10所示p6最大吸收波长约为400nm。
向p16的dmso溶液(浓度为10-5m)中加入2.3μl三氟乙酸(1000eq),测定加酸前后时间分辨的紫外可见吸收光谱,结果显示p16的最大吸收波长处的吸光度没有随着酸化时间的增长而增加,表明p16分子仍然具有耐酸的特性。
将snap-微管蛋白融合质粒通过标准的基因工程方法转染u2os细胞,培养24小时后取10ulp16的dmso溶液(浓度为10-3m)加入1ml培养基中染色培养u2os细胞1小时,随后弃去培养基溶液并加入1mlpbs缓冲溶液清洗掉未染色的多余染料,反复清洗5-8次后,加入1ml4%的多聚甲醛pbs溶液固定细胞10分钟后,用pbs溶液洗涤3-4次即可置于storm显微镜下,选择常用的405nm激光作为激活光,561nm激光作为激发光,通过随机光激活以及重叠重构技术即可得到超分辨图像,如图11所示,超分辨2d-storm成像得到的微管图像比宽场荧光图像分辨率大大提高。
实施例17
当r1=r2=r3=r4=c2h5,r5=ch3co,x=o,y=h,z=1-(4-苄基鸟嘌呤)-6-(4-苯乙炔基)萘酰亚胺(c33h22n6o3)时,其分子(p17)合成路线和产物结构如下:
合成步骤及表征:p12(0.39g,0.5mmol)和bg-nh2(0.19g,1.5mmol)混合于无水乙醇(10ml),升温至78℃回流,搅拌10小时后减压蒸除溶剂。粗产物通过柱色谱(硅胶,乙酸乙酯/石油醚,1:2v/v)分离提纯得到黄色粉末产物p17(0.44g,84%)。
对黄色粉末产物进行了核磁和质谱的表征:1hnmr(400mhz,dmso)δ12.40(s,1h),10.42(s,1h),8.71(d,j=8.5hz,1h),8.55(d,j=6.9hz,1h),8.45(d,j=7.7hz,1h),8.33(d,j=8.2hz,1h),7.99(d,j=7.7hz,1h),7.92(t,j=7.8hz,1h),7.77(s,1h),7.60(d,j=8.6hz,2h),7.52(t,j=7.9hz,1h),7.43(d,j=8.2hz,2h),7.38(d,j=8.2hz,2h),7.09(d,j=8.6hz,2h),6.71–6.64(m,3h),6.39(dd,j=9.0,2.4hz,2h),6.32(d,j=2.3hz,2h),6.25(s,1h),5.75(s,1h),5.42(s,2h),5.24(s,2h),3.29(dd,j=13.9,6.8hz,8h),2.24(s,3h),1.06(t,j=6.9hz,12h)。13cnmr(101mhz,dmso)δ169.90,169.09,164.28,163.98,160.59,155.01,153.07,149.54,138.74,138.07,137.97,136.04,133.30,132.57,131.89,131.31,129.61,129.30,128.63,128.47,127.30,126.19,123.45,122.80,119.92,118.62,118.20,115.43,109.27,105.82,99.34,98.16,87.62,67.76,55.90,44.62,25.61,13.38。lc-ms(esi):m/z:计算值:1046.4228;实验值:524.2137[m+2h]2+/2。
经上述检测,鉴定其结构为p17所示。
将产物p17溶于dmso中配成溶液(浓度为10-5m),测试溶液的紫外可见吸收光谱,如图10所示p17最大吸收波长约为400nm。
向p17的dmso溶液(浓度为10-5m)中加入2.3μl三氟乙酸(1000eq),测定加酸前后时间分辨的紫外可见吸收光谱,结果显示p17的最大吸收波长处的吸光度没有随着酸化时间的增长而增加,表明p17分子仍然具有耐酸的特性。
将snap-微管蛋白融合质粒通过标准的基因工程方法转染u2os细胞,培养24小时后取10ulp17的dmso溶液(浓度为10-3m)加入1ml培养基中染色培养u2os细胞1小时,随后弃去培养基溶液并加入1mlpbs缓冲溶液清洗掉未染色的多余染料,反复清洗5-8次后,加入1ml4%的多聚甲醛pbs溶液固定细胞10分钟后,用pbs溶液洗涤3-4次即可置于storm显微镜下,选择常用的405nm激光作为激活光,561nm激光作为激发光,通过随机光激活以及重叠重构技术即可得到超分辨图像,如图12所示,超分辨3d-storm成像得到的微管图像比宽场荧光图像分辨率大大提高。
- 还没有人留言评论。精彩留言会获得点赞!