一种氯代苯氧羧酸胺盐的制备方法与流程
本发明涉及除草剂制备技术领域,尤其涉及一种氯代苯氧羧酸胺盐的制备方法。
背景技术
目前,氯代苯氧羧酸胺盐的制备方法主要包括以下几步:
1)以酚为主要原料,经氯化制得氯代酚。此步产出的氯代酚具有极难闻的刺激性气味,导致生产现场环境极差,而且氯化选择性较差。
2)氯代酚在碱性条件下与氯代羧酸缩合,反应液经过酸化、过滤得到氯代苯氧羧酸湿料,烘干后得氯代苯氧羧酸。此步氯代酚中的二氯酚或多氯酚在缩合过程中,会发生两分子间的缩合,产生极难降解的剧毒物质--二噁英,而且生产的氯代苯氧羧酸中也含有二噁英,给环境和生产人员的健康带来了极大的风险。
3)以氯代苯氧羧酸、胺为原料,经酸碱中和反应合成氯代苯氧羧酸胺盐。在此步氯代苯氧羧酸中含有的二噁英会进入氯代苯氧羧酸胺盐中,并随着氯代苯氧羧酸胺盐的使用进入植物体、空气、土壤和水源,并随着食物链富集,进而造成更加严重的环境危害。
上述制备过程中,氯化选择性差、后处理工艺会造成有效成分损失,产品的收率偏低。同时在使用酚为原料经氯化、缩合合成氯代苯氧羧酸时,会产出大量的含有羟基羧酸和废盐的废水,以及大量含有氯代酚、氯代苯氧羧酸的危废,三废处理压力大、处理成本高。
在将缩合所得的氯代苯氧羧酸盐酸化,然后铵化成盐的过程中,不仅产生大量的高盐废水,还会造成有效成分的损失,而且流程长增加了设备投入和生产成本。
现有的氯代苯氧羧酸胺盐的合成工艺已经十分落后,随着环保意识、环保标准的不断提高,老旧落后的工艺已经严重制约着氯代苯氧羧酸胺盐的合成产业的良性发展和可持续发展,开发一种先进的合成工艺迫在眉睫。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种氯代苯氧羧酸胺盐的制备方法,具有较高的收率和纯度,且安全环保。
为解决以上技术问题,本发明提供了一种氯代苯氧羧酸胺盐的制备方法,包括以下步骤:
s1)苯氧羧酸酯在催化剂a和催化剂b的作用下,和氯化剂进行2位和/或4位的选择性氯化反应,得到氯代苯氧羧酸酯;
所述催化剂a为路易斯酸;
所述催化剂b为c5~22的噻唑、异噻唑、噻吩或它们的卤代衍生物;
s2)氯代苯氧羧酸酯和胺进行氨解反应,得到氯代苯氧羧酸胺盐。
本发明中,所述苯氧羧酸酯具有以下式ⅰ~式ⅳ任一结构:
其中,r1优选为c1~c3的亚烷基,进一步的,优选为亚甲基(-ch2-),甲基亚甲基(-ch(ch3)-),亚乙基(-ch2-ch2-)或亚丙基(-ch2-ch2-ch2-)。
r优选为c1~c10的烷基或c3~c10的环烷基,更优选为甲基、乙基、丙基、异丙基、丁基、异丁基、辛基、异辛基、环丙基或环己基。
对应的氯化产物结构如下:
所述催化剂a为路易斯酸;优选为sncl4、mgcl2、fecl3、alcl3、bf3、zncl2、ticl4、sbf5、al2o3、fe2o3、tio2、pb(oac)2、zn(oac)2和al2o(oac)4中的一种或多种;更优选为mgcl2、fecl3、zncl2、sbf5、tio2和pb(oac)2中的一种或多种,进一步优选为fecl3、tio2和pb(oac)2中的一种或多种。
所述催化剂b为c5~22的噻唑、异噻唑、噻吩或它们的卤代衍生物;优选为噻唑、2-乙基噻唑、2,5-二氯噻唑、4-甲基噻唑、2-叔丁基噻唑、异噻唑、4,5-二甲基异噻唑、5-氯异噻唑、2,4,5-三叔丁基异噻唑、噻吩、2-甲基噻吩、2,5-二甲基噻吩、3-氯噻吩、3,4-二氯噻吩和2,3,4-三氯噻吩中的一种或多种;更优选为2-乙基噻唑、2,5-二氯噻唑、2,4,5-三叔丁基异噻唑、4,5-二甲基异噻唑、3,4-二氯噻吩和2,3,4-三氯噻吩中的一种或多种;进一步优选为2,4,5-三叔丁基异噻唑和2,3,4-三氯噻吩中的一种或多种。
所述催化剂a的用量优选为苯氧羧酸酯重量的0.05%~1.0%,更优选为0.25%~1.0%,进一步优选为0.5%~1.0%。
所述催化剂b的用量优选为苯氧羧酸酯重量的0.05%~1.0%,更优选为0.2%~0.8%,进一步优选为0.3%~0.5%。
所述催化剂a的用量不宜过少,否则会导致氯化选择性出现大幅下降,但用量过多除了不经济外还会增加产品分离的难度;催化剂b的用量不宜过少,否则会导致反应不发生或反应缓慢,用量过多不仅不经济还会导致选择性有所降低,同时还会增加产品分离难度。
所述氯化剂可以为苯酚氯化反应的一般氯化剂,优选为氯气、亚硫酰氯或硫酰氯,更优选为氯气或硫酰氯。
所述2位和/或4位的选择性氯化反应,指2位的单取代反应,4位的单取代反应,或2位和4位的双取代反应。
当所述选择性氯化反应为在2位或4位进行单取代反应时,如苯氧羧酸酯的结构如式ⅰ~式ⅳ所示,所述苯氧羧酸酯与氯化剂的摩尔比优选为1:(0.99~1.2),更优选为1:(1~1.1),进一步优选为1:(1.01~1.03)。
当所述选择性氯化反应为二取代反应时,如苯氧羧酸酯的结构如式ⅰ所示,产物为2,4-二氯苯氧羧酸酯,所述苯氧羧酸酯与氯化剂的摩尔比优选为1:(1.98~2.4),更优选为1:(2~2.2),进一步优选为1:(2.02~2.06)。
氯化剂的用量不宜过少,否则会使原料的未转偏高,但是用量过多又会导致较多的过氯化产物的生成,对反应不利。
所述选择性氯化反应的温度优选为-20~100℃,更优选为-20~60℃,进一步优选为-20~20℃。所述反应的时间优选为0.2~1h。
该反应温度可以同时保持较高的反应活性和氯化选择性。
所述选择性氯化反应结束后,优选进行减压蒸馏,收集相应沸程的馏分即可得到氯代苯氧羧酸酯。
氯代苯氧羧酸酯和胺进行氨解反应,生成氯代苯氧羧酸酰胺中间体,然后在高温下与水反应进而生成氯代苯氧羧酸胺盐。
具体的,向氯代苯氧羧酸酯中加入一定比例的胺,于一定温度下进行氨解反应得氯代苯氧羧酸胺盐和醇(roh),蒸馏回收醇roh,余下物料即为氯代苯氧羧酸胺盐。
所述胺的分子式为nr2r3r4,其中,r2、r3和r4独立的优选为h或c1~c5的烷基,且r2、r3和r4的碳原子总数≤5。r2、r3和r4可以相同或不同。
所述胺优选为氨、一甲胺、二甲胺、三甲胺、乙胺、二乙胺、正丙胺、异丙胺、正丁胺、异丁胺、叔丁胺、正戊胺、异戊胺、新戊胺等,进一步优选为二甲胺。
氯代苯氧羧酸酯与胺的摩尔比优选为1:(1~1.2),更优选为1:(1~1.12),进一步优选为1:(1.02~1.06)。
所述氨解反应的温度优选为100~180℃,更优选为120~160℃。
在本发明的某些具体实施例中,得到的氯代苯氧羧酸胺盐结构如下:
本发明制备的氯代苯氧羧酸胺盐可以直接作为除草剂产品,也可直接加入助剂做成各种除草剂制剂。
与现有技术相比,本发明提供了一种氯代苯氧羧酸胺盐的制备方法,包括以下步骤:s1)苯氧羧酸酯在催化剂a和催化剂b的作用下,和氯化剂进行2位和/或4位的选择性氯化反应,得到氯代苯氧羧酸酯;所述催化剂a为路易斯酸;所述催化剂b为c5~22的噻唑、异噻唑、噻吩或它们的卤代衍生物;s2)氯代苯氧羧酸酯和胺进行氨解反应,得到氯代苯氧羧酸胺盐。
本发明以苯氧羧酸酯为原料,经选择性氯化合成氯代苯氧羧酸酯,然后氨解得到相应的胺盐,与现有合成技术相比,工艺路线得到了极大的简化,有效的避免了具有难闻气味的氯代酚的生产和使用,从根本上杜绝了剧毒的二噁英的产生,同时中间产物和产品中也不会含有氯代酚,极大的改善了产品品质和生产现场的操作环境。
合成过程中不仅不会产生含有羟基羧酸和金属氯化物的废水和含氯代酚、氯代苯氧羧酸的危废,还简化了后处理,有效杜绝了高cod、高盐废水的产生,极大地降低了三废处理量、三废处理难度和处理成本。
最终本发明通过对工艺路线的重新设计,对催化剂和氯化剂的精细筛选,有效降低了能耗,提高了氯化选择性同时避免了有效成分的损失,所得氯代苯氧羧酸胺盐的收率可达99%以上。与现有工艺相比,有效提高了选择性,提高了产品品质,杜绝了高盐废水的产出,避免了氯代苯氧羧酸烘干及使用造成的粉尘危害,节约了能源,降低了设备投入。
本发明制备的氯代苯氧羧酸胺盐的杂质及杂质含量如下表1所示:
表1本发明制备的氯代苯氧羧酸胺盐的杂质及杂质含量汇总
附图说明
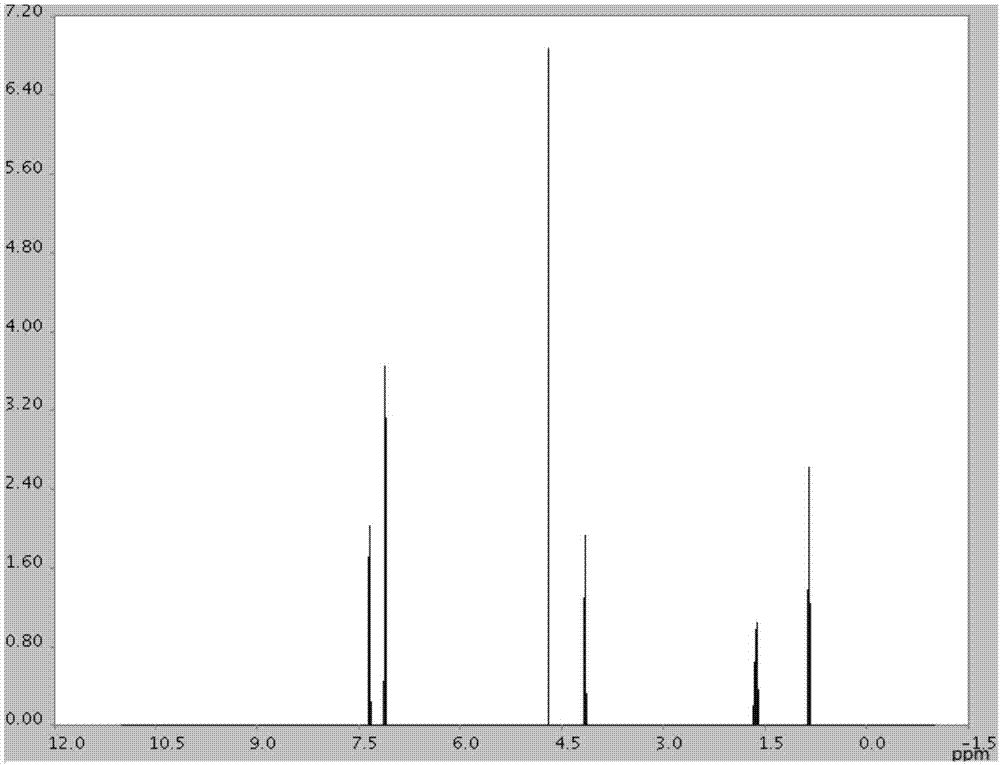
图1为本发明实施例2所得4-氯苯氧乙酸丙酯的核磁共振氢谱图。
具体实施方式
为了进一步说明本发明,下面结合实施例对本发明提供的氯代苯氧羧酸胺盐的制备方法进行详细描述。
实施例1
向500ml四口瓶中依次加入167.87g99%的苯氧乙酸甲酯(1mol)、1.26g99%的氯化锌和1.68g99%的2,5-二氯噻唑,于-20℃下通入154.69g99%的氯气(2.16mol),通完保温反应30min,于1kpa压力下蒸馏并收集140~150℃的馏分,得2,4-二氯苯氧乙酸甲酯236.21g,含量98.8%,收率以苯氧乙酸甲酯计99.28%。
经检测,杂质含量如下:4-氯苯氧乙酸甲酯含量0.03%,2,6-二氯苯氧乙酸甲酯0.09%,2,4,6-三氯苯氧乙酸甲酯含量0.16%,2,5-二氯噻唑含量0.13%。
向蒸馏所得2,4-二氯苯氧乙酸甲酯中加入118.63g40%的二甲胺,于130℃下反应并蒸出生成的醇,得2,4-二氯苯氧乙酸二甲胺盐319.14g,含量82.5%,收率以苯氧乙酸甲酯计98.88%。
比较例1
向500ml四口瓶中依次加入167.87g99%的苯氧乙酸甲酯(1mol)、1.26g99%的氯化锌,于-20℃下通入154.69g99%的氯气(2.16mol),通完保温反应30min,于1kpa压力下蒸馏并收集140~150℃的馏分,得2,4-二氯苯氧乙酸甲酯215.52g,含量98.5%,收率以苯氧乙酸甲酯计90.31%。
向蒸馏所得2,4-二氯苯氧乙酸甲酯中加入98.16g40%的二甲胺,于130℃下反应并蒸出生成的醇,得2,4-二氯苯氧乙酸二甲胺盐291.24g,含量81.6%,收率以苯氧乙酸甲酯计89.25%。
比较例2
向500ml四口瓶中依次加入167.87g99%的苯氧乙酸甲酯(1mol)、1.68g99%的2,5-二氯噻唑,于-20℃下通入154.69g99%的氯气(2.16mol),通完保温反应30min,于1kpa压力下蒸馏并收集140~150℃的馏分,得2,4-二氯苯氧乙酸甲酯192.81g,含量98.3%,收率以苯氧乙酸甲酯计80.63%。
向蒸馏所得2,4-二氯苯氧乙酸甲酯中加入96.34g40%的二甲胺,于130℃下反应并蒸出生成的醇,得2,4-二氯苯氧乙酸二甲胺盐264.16g,含量80.5%,收率以苯氧乙酸甲酯计79.86%。
实施例2
向500ml四口瓶中依次加入196.21g99%的苯氧乙酸丙酯(1mol)、0.29g99%的氯化铁和0.69g99%的2-乙基噻唑,于30℃下滴加入137.70g99%的硫酰氯(1.01mol),滴加完毕保温反应30min,于1kpa压力下蒸馏并收集145~155℃的馏分,得4-氯苯氧乙酸丙酯229.01g,含量98.9%,收率以苯氧乙酸丙酯计99.04%。
向蒸馏所得4-氯苯氧乙酸丙酯中加入129.51g40%的二甲胺,于120℃下反应并蒸出生成的醇,得4-氯苯氧乙酸二甲胺盐291.58g,含量78.4%,收率以苯氧乙酸丙酯计98.64%。
采用核磁共振对制备的4-氯苯氧乙酸丙酯进行检测,结果见图1,图1为4-氯苯氧乙酸丙酯的核磁共振氢谱图。
实施例3
向500ml四口瓶中依次加入196.21g99%的苯氧丁酸甲酯(1mol)、1.08g99%的四氯化钛和0.88g99%的2,3,4-三氯噻吩,于0℃下通入157.56g99%的氯气(2.2mol),通完保温反应30min,于1kpa压力下蒸馏并收集150~160℃的馏分,得2,4-二氯苯氧丁酸甲酯264.04g,含量98.8%,收率以苯氧丁酸甲酯计99.14%。
向蒸馏所得2,4-二氯苯氧丁酸甲酯中加入72.94g25%的氨水,于100℃下反应并蒸出生成的醇,得2,4-二氯苯氧丁酸铵盐303.00g,含量86.7%,收率以苯氧丁酸甲酯计98.74%。
实施例4
向500ml四口瓶中依次加入196.21g99%的2-苯氧基丙酸乙酯(1mol)、0.10g99%的氯化铝和0.29g99%的2,4,5-三叔丁基异噻唑,于50℃下通入85.94g99%的氯气(1.2mol),通完保温反应30min,于1kpa压力下蒸馏并收集145~155℃的馏分,得2-(4-氯苯氧基)丙酸乙酯229.13g,含量99.1%,收率以2-苯氧基丙酸乙酯计99.29%。
向蒸馏所得2-(4-氯苯氧基)丙酸乙酯中加入176.10g40%的异丙胺,于160℃下反应并蒸出生成的醇,得2-(4-氯苯氧基)丙酸异丙胺盐347.25g,含量74.0%,收率以2-苯氧基丙酸乙酯计98.89%。
实施例5
向500ml四口瓶中依次加入259.34g99%的2-氯苯氧丁酸环丙酯(1mol)、0.91g99%的二氧化钛和1.43g99%的2-甲基噻吩,于60℃下滴加入140.42g99%的硫酰氯(1.03mol),通完保温反应30min,于1kpa压力下蒸馏并收集160~170℃的馏分,得2,4-二氯苯氧丁酸环丙酯291.72g,含量99.2%,收率以2-氯苯氧丁酸环丙酯计99.38%。
向蒸馏所得2,4-二氯苯氧丁酸环丙酯中加入185.38g40%的正丁胺,于150℃下反应并蒸出生成的醇,得2,4-二氯苯氧丁酸正丁胺盐415.32g,含量76.8%,收率以2-氯苯氧丁酸环丙酯计98.98%。
实施例6
向500ml四口瓶中依次加入196.21g99%的2-苯氧基丙酸乙酯(1mol)、0.49g99%的醋酸铅和0.10g99%的3,4-二氯噻吩,于40℃下滴加入237.92g99%的硫酰氯(1.98mol),滴完保温反应30min,于1kpa压力下蒸馏并收集150~160℃的馏分,得2-(2,4-二氯苯氧基)丙酸乙酯263.16g,含量99.2%,收率以2-苯氧基丙酸乙酯计99.21%。
向蒸馏所得2-(2,4-二氯苯氧基)丙酸乙酯中加入112.95g40%的二甲胺,于110℃下反应并蒸出生成的醇,得2-(2,4-二氯苯氧基)丙酸二甲胺盐329.92g,含量83.9%,收率以2-苯氧基丙酸乙酯计98.81%。
实施例7
向500ml四口瓶中依次加入196.21g99%的2-(4-氯苯氧基)丙酸丙酯(1mol)、2.45g99%的氧化铝和2.08g99%的2,5-二甲基噻吩,于100℃下通入85.94g99%的硫酰氯(1.2mol),通完保温反应30min,于1kpa压力下蒸馏并收集155~165℃的馏分,得2-(2,4-二氯苯氧基)丙酸丙酯276.52g,含量99.2%,收率以2-(4-氯苯氧基)丙酸丙酯计98.97%。
向蒸馏所得2-(2,4-二氯苯氧基)丙酸丙酯中加入86.07g40%的一甲胺,于100℃下反应并蒸出生成的醇,得2-(2,4-二氯苯氧基)丙酸一甲胺盐300.17g,含量87.4%,收率以2-(4-氯苯氧基)丙酸丙酯计98.57%。
实施例8
向500ml四口瓶中依次加入196.21g99%的苯氧乙酸异丙酯(1mol)、0.88g99%的氯化镁和0.49g99%的2,5-二氯噻唑,于70℃下滴入275.39g99%的硫酰氯(2.02mol),通完保温反应30min,于1kpa压力下蒸馏并收集150~160℃的馏分,得2,4-二氯苯氧乙酸异丙酯263.64g,含量98.9%,收率以苯氧乙酸异丙酯计99.09%。
向蒸馏所得2,4-二氯苯氧乙酸异丙酯中加入188.46g40%的二乙胺,于180℃下反应并蒸出生成的醇,得2,4-二氯苯氧乙酸二乙胺盐389.08g,含量74.6%,收率以苯氧乙酸异丙酯计98.69%。
实施例9
向500ml四口瓶中依次加入182.04g99%的2-甲基苯氧乙酸甲酯(1mol)、1.18g99%的氧化铁和1.37g99%的4,5-二甲基异噻唑,于80℃下滴入140.42g99%的硫酰氯(1.03mol),滴完保温反应30min,于1kpa压力下蒸馏并收集140~150℃的馏分,得4-氯-2-甲基苯氧乙酸甲酯214.70g,含量99.3%,收率以2-甲基苯氧乙酸甲酯计99.32%。
向蒸馏所得4-氯-2-甲基苯氧乙酸甲酯中加入229.46g40%的正戊胺,于160℃下反应并蒸出生成的醇,得4-氯-2-甲基苯氧乙酸正戊胺盐406.87g,含量82.1%,收率以2-甲基苯氧乙酸甲酯计99.09%。
实施例10
向500ml四口瓶中依次加入238.73g99%的2-甲基苯氧丁酸异丙酯(1mol)、2.03g99%的四氯化锡和1.55g99%的4-甲基噻唑,于20℃下滴入118.96g99%的亚硫酰氯(0.99mol),滴完保温反应30min,于1kpa压力下蒸馏并收集140~150℃的馏分,得4-氯-2-甲基苯氧丁酸异丙酯271.57g,含量99.2%,收率以2-甲基苯氧丁酸异丙酯计99.49%。
向蒸馏所得4-氯-2-甲基苯氧丁酸异丙酯中加入114.39g40%的二甲胺,于150℃下反应并蒸出生成的醇,得4-氯-2-甲基苯氧丁酸二甲胺盐296.73g,含量82.1%,收率以2-甲基苯氧丁酸异丙酯计99.09%。
由上述实施例及比较例可知,本发明提供的制备方法具有较高的收率和纯度。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!