用于制备bictegravir的中间体及其制备方法与流程
本发明涉及医药技术领域,尤其涉及用于制备bictegravir的中间体及其制备方法。
背景技术
bictegravir(gs-9883)是由gilead公司研制的一类新型整合酶抑制剂,与之前研制的整合酶抑制剂不同,bictegravir只需每日使用一次,且无需增效剂cobicistat,目前在临床试验第3期,即taf/ftc/bic方案的研究。以往的研究表明,bictegravir在健康人群中耐受性良好,可抑制肾小管转运蛋白,降低肌酐水平,导致肾小球滤过率下降,但并不损伤肾功能。此外,bictegravir比其他整合酶抑制剂具有更高的耐药屏障,且与其他药物之间的相互作用较少。
波士顿2017croi大会上公布了正在开展的一项2期临床安慰剂双盲对照研究结果,该研究旨在比较bictegravir与dolutegravir用作一线治疗方案的疗效。研究结果表明,bictegravir方案与dolutegravir(tivcay)方案的疗效相似,且具有良好的安全性和耐受性。
目前在欧洲和美国的治疗指南中,bictegravir被推荐作为一线治疗的首选方案。bictegravir是gilead自taf后,在hiv药物领域又一即将投入市场的重磅药,预计将为gilead稳住hiv药物市场占有率的垄断地位。
wo2015195656公开了一种bictegravir的制备方法,其合成路线如下:
该路线虽然较短,但是成本很高,单一构型的原料极其昂贵,如果选用相对便宜的消旋体为原料,手性拆分只能在路线的后期进行,且收率较低;拜耳-维立格氧化中用到的过氧化物属于易爆品,放大过程中具有一定的危险性,不适合工业化生产。
wo2015195656还公开了一种bictegravir的制备方法,其合成路线如下:
该路线虽然原料廉价易得,但是,第一步环氧化收率较低;第二步反应需要用到不可回收的均相钯催化剂,且收率较低;得到的产物为消旋体,后期手性拆分的成本很高。
鉴于bictegravir良好的药用前景,因此需要开发一种经济、安全的bictegravir的制备方法。
技术实现要素:
本发明要解决的技术问题是:提供一种制备bictegravir的中间体。
在本发明的第一方面,提供了一种制备bictegravir的中间体,其结构如式(ivb)所示:
其中r基团定义如下:rcooh为手性羧酸,所述手性羧酸选自手性羟基酸、手性氨基酸、手性碳水化合物或手性萜类化合物;其中手性羟基酸选自乳酸,酒石酸,苹果酸;手性氨基酸选自丙氨酸,精氨酸,门冬氨酸,半胱氨酸,谷氨酸,异亮氨酸,亮氨酸,赖氨酸,甲硫氨酸,苯甘氨酸,脯氨酸,焦谷氨酸,丝氨酸,色氨酸,苏氨酸,苯丙氨酸;手性碳水化合物选自抗坏血酸,氯醛酸,半乳糖酸,葡庚糖酸,葡糖酸,异抗坏血酸,葡糖二酸;手性萜类衍生物,如樟脑酸。
在本发明的第二方面,提供了一种制备如式(ivb)所示中间体的方法,包括如下步骤:
(1)式(i)化合物与羟胺缩合得到式(ii)化合物;
(2)式(ii)化合物经过氧化、不对称diels-alder反应得到式(iii)化合物;
(3)式(iii)化合物经过催化氢化得到式(ivb)化合物;
本发明的第三方面,提供了一种制备如式(ⅵ)所示化合物的方法,
包括如下步骤:
(1)式ivb化合物经过切断酰胺键反应得到式vb化合物
(2)式vb化合物经过boc保护得到式vib化合物
(3)式vib经过催化氢化得到式v化合物
(4)式v化合物经过脱boc反应,成盐得到式vi化合物
本发明的第四方面,提供了另一种制备bictegravir的中间体,其结构如式(ivc)所示:
其中r基团定义如下:rcooh为手性羧酸,所述手性羧酸选自手性羟基酸、手性氨基酸、手性碳水化合物或手性萜类化合物;其中手性羟基酸选自乳酸,酒石酸,苹果酸;手性氨基酸选自丙氨酸,精氨酸,门冬氨酸,半胱氨酸,谷氨酸,异亮氨酸,亮氨酸,赖氨酸,甲硫氨酸,苯甘氨酸,脯氨酸,焦谷氨酸,丝氨酸,色氨酸,苏氨酸,苯丙氨酸;手性碳水化合物选自抗坏血酸,氯醛酸,半乳糖酸,葡庚糖酸,葡糖酸,异抗坏血酸,葡糖二酸;手性萜类衍生物,如樟脑酸。
本发明的第五方面,提供了一种制备式(ivc)所示中间体的方法,包括如下步骤:
(1)式i化合物与羟胺缩合得到式ii化合物;
(2)式ii化合物经过氧化、不对称diels-alder反应得到式iii化合物;
(3)式iii化合物经过催化氢化得到式ivc化合物;
本发明的第六方面,提供了一种制备如式(ⅵ)所示化合物的方法,包括如下步骤:
(1)式ivc化合物经过切酰胺键反应和boc保护反应得到式v化合物;
(2)式v化合物经过脱boc反应,成盐得到式vi化合物;
本发明的第七方面,提供了一种制备如式(ⅵ)所示化合物的方法,包括如下步骤:
(1)式iii化合物经过切断酰胺键反应得到式iva化合物;
(2)式iva化合物经过催化氢化和boc保护得到式v化合物;
(3)式v化合物经过脱boc反应,成盐得到式vi化合物;
其中r基团定义如下:rcooh为手性羧酸,所述手性羧酸选自手性羟基酸、手性氨基酸、手性碳水化合物或手性萜类化合物;其中手性羟基酸选自乳酸、酒石酸或苹果酸;手性氨基酸选自丙氨酸、精氨酸、门冬氨酸、半胱氨酸、谷氨酸、异亮氨酸、亮氨酸、赖氨酸、甲硫氨酸、苯甘氨酸、脯氨酸、焦谷氨酸、丝氨酸、色氨酸、苏氨酸或苯丙氨酸;手性碳水化合物选自抗坏血酸、氯醛酸、半乳糖酸、葡庚糖酸、葡糖酸、异抗坏血酸或葡糖二酸;手性萜类衍生物,选自樟脑酸。
优选的,步骤(1)可以依靠底物诱导、手性催化或者两者协同的方式进行。手性催化剂包括双噁唑啉类催化剂、手性酰氧基硼烷催化剂、手性镧系催化剂,双磺酰胺类催化剂(corey催化剂),narasaka催化剂等。
优选的,氧化所述化合物ii的氧化方式为斯文氧化或者高碘化物氧化;其中,高碘化物包括:高碘酸钠、高碘酸钾、四乙基高碘酸胺等。
优选的,本申请中所涉及的催化氢化的催化剂包括雷尼镍、钯碳、氢氧化钯碳、二氧化铂、铑碳或钌碳等。
优选的,本申请中所涉及的切断酰胺键的方法包括水解、氨解、肼解或者硼氢化物及其衍生物,硼氢化物/金属盐体系;其中,所述硼氢化物包括:硼氢化锂、硼氢化钠及硼氢化钾;所述金属盐体系包括:碱土金属卤化物、镧系金属卤化物及过渡金属卤化物;所述硼氢化物衍生物包括氰基硼氢化钠、三异丙氧基硼氢化钾或三乙基硼氢化锂。
本发明通过手性辅剂的诱导作用,大幅度地提高了diels-alder反应的立体选择性,制得较高手性纯度的公共中间体(iii);n-o键的切断和双键的还原均使用催化氢化,绿色环保;路线反应条件温和,且收率比现有的制备方法高,经济有效,适于大规模的工业化生产。
附图说明
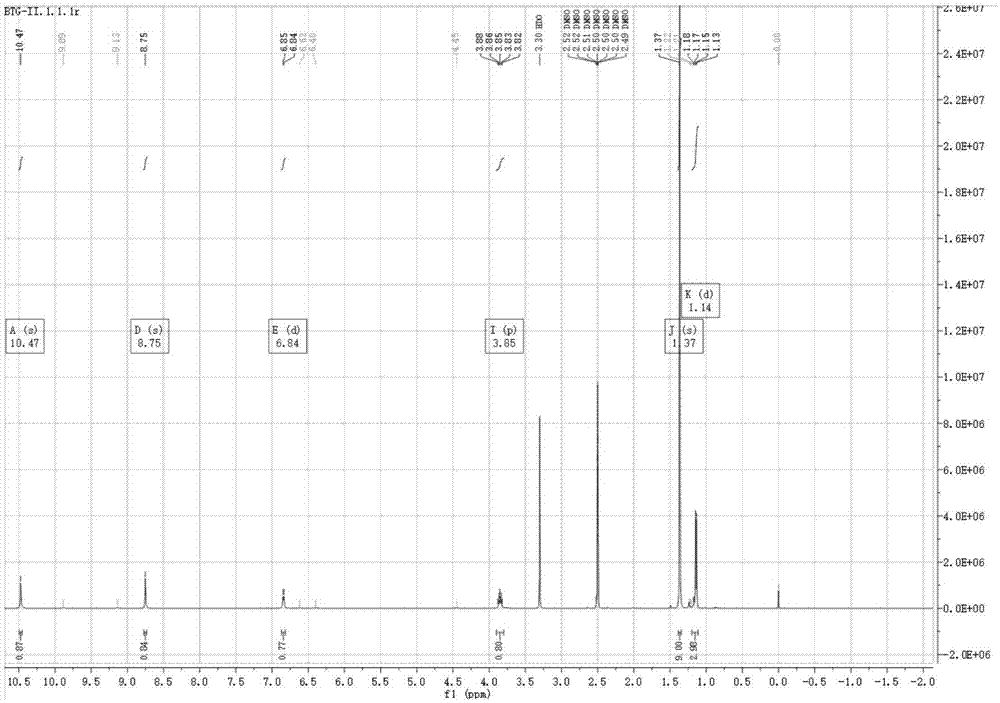
图1是式ii化合物的氢谱图;
图2是式iii化合物的氢谱图;
图3是式v化合物的氢谱图;
图4是式vi化合物的氢谱图;
图5是式ivb化合物的氢谱图;
图6是式vib化合物的氢谱图;
图7是式ivc化合物的氢谱图。
具体实施方式
以下结合实例说明本发明,但不限制本发明。在本领域内,技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案内。
实施例1:
式i化合物与羟胺缩合得到式ii化合物;其中rcooh为d-丙氨酸。
将147.8g式i化合物溶于450ml四氢呋喃中,加入79.5g三乙胺,降温至0-5℃,滴加106.5g氯甲酸异丁酯,大量固体析出,保温2h;将81.75g盐酸羟胺溶于750ml甲醇中,降温至0-5℃,分批加入氢氧化钠,大量固体析出,保温2h,加入步骤一的滤液,继续搅拌2h。抽滤,滤液减压浓缩,加入1l乙酸乙酯,白色固体析出,抽滤,滤饼用200ml乙酸乙酯洗涤,浓缩至干。750ml二氯甲烷打浆,抽滤,45℃鼓风干燥,得到137.2g白色固体,收率86%。1hnmr(500mhz,dmso-d6)δ10.47(s,1h),8.75(s,1h),6.84(d,j=7.7hz,1h),3.85(p,j=7.3hz,1h),1.37(s,9h),1.14(d,j=7.1hz,3h).
实施例2:
式ii化合物经过氧化、不对称diels-alder反应得到式iii化合物;其中rcooh为d-丙氨酸。
将12.5g式ii化合物溶于300ml甲醇和100ml水中,降温至0-5℃。加入12.6g高碘酸钠和14ml环戊二烯,溶液颜色迅速变深,大量淡黄色固体析出,温度稍有上升,控制温度不超过5℃,搅拌30min后,加入10ml环戊二烯和7.6g高碘酸钠,保温反应1h。旋干,柱层析得到11.9g白色固体,收率73%。
1hnmr(500mhz,dmso-d6)δ6.97(s,1h),6.55–6.50(m,1h),6.43(s,1h),5.45(s,1h),5.28(s,1h),4.15(s,1h),1.85(d,j=9.1hz,1h),1.78(d,j=8.9hz,1h),1.36(s,9h),0.91(s,3h).
实施例3:
式iii化合物经过切断酰胺键反应得到式iva化合物,式iva化合物经过催化氢化和boc保护得到式v化合物;其中式iii化合物中rcooh为d-丙氨酸;
将10g式iii化合物溶于60ml甲醇和20ml水中,室温下加入2.5g氢氧化锂,搅拌2h后,加入8.1gboc酸酐,室温搅拌1h。旋干,加入20ml水,30ml二氯甲烷萃取2次,无水硫酸钠干燥。旋干,柱层析,得到3.6g油状物式v化合物,收率48%。
1hnmr(500mhz,dmso-d6)δ6.63(d,j=7.7hz,1h),4.54(d,j=4.5hz,1h),4.06-3.97(m,1h),3.70(h,j=7.3hz,1h),2.01(dt,j=13.5,6.9hz,1h),1.79-1.66(m,1h),1.63(ddt,j=13.8,10.0,6.5hz,1h),1.50(pd,j=8.5,4.6hz,2h),1.37(s,9h),1.31(ddd,j=12.8,7.1,5.2hz,1h).
实施例4:
式v化合物经过脱boc反应,成盐得到式vi化合物
将10g式v化合物溶于20ml二氧六环中,加入50ml盐酸二氧六环(4m),室温搅拌2h。旋干反应液,100ml乙腈打浆,抽滤,滤饼用100ml乙腈漂洗,鼓风干燥,得到6.4g白色固体,收率95%。
1hnmr(500mhz,dmso-d6)δ8.10(s,3h),4.98(s,1h),4.11(p,j=5.0hz,1h),3.47–3.37
(m,1h),2.07(ddd,j=13.7,7.8,5.9hz,1h),1.96-1.85(m,1h),1.81-1.66(m,2h),1.69-1.61(m,1h),1.58(dt,j=13.7,5.1hz,1h).
实施例5:
式iii化合物经过催化氢化得到式ivb化合物,其中rcooh为d-丙氨酸。
将10g式(iii)化合物溶于100ml甲醇中,加入1gpd/c(5%),氢气置换3次,室温搅拌8h,抽滤,适量甲醇漂洗滤饼。旋干,柱层析,得到9g油状物,收率89%。
1hnmr(500mhz,dmso-d6)δ6.88(s,1h),4.85(s,1h),4.70(s,1h),4.21(p,j=7.3hz,1h),1.82(q,j=10.4,10.0hz,1h),1.70(d,j=9.9hz,4h),1.65(s,1h),1.36(s,9h),1.14(d,j=7.2hz,3h).
实施例6:
式ivb化合物经过切断酰胺键反应得到式vb化合物,式vb化合物经过boc保护得到式vib化合物,其中rcooh为d-丙氨酸。
将10g式ivb化合物溶于60ml甲醇和20ml水中,室温下加入2.5g氢氧化锂,搅拌7h后,旋至不滴,加入20ml水,30ml二氯甲烷萃取三次。合并有机层,加入6.6gboc酸酐,室温搅拌30min。旋干反应液,柱层析得到6.2g油状物,收率82.5%。
1hnmr(500mhz,dmso-d6)δ4.66(d,j=2.0hz,1h),4.41(s,1h),1.76-1.57(m,6h),1.40(s,9h).
实施例7:
式vib经过催化氢化得到式v化合物
将10g式(vib)化合物溶于100ml甲醇中,加入1gpd/c(5%),氢气置换3次,室温搅拌48h,抽滤,适量甲醇漂洗滤饼。旋干,柱层析,得到9.2g油状物,收率91%。
实施例8:
式iii化合物经过催化氢化反应得到式ivc化合物,其中rcooh为d-丙氨酸。
将10g式(iii)化合物溶于100ml甲醇中,加入1gpd/c(5%),氢气置换3次,60℃,8公斤压力反应8h,抽滤,适量甲醇漂洗滤饼。旋干,柱层析,得到9.3g油状物,收率92%。1hnmr(500mhz,dmso-d6)δ7.62(d,j=7.8hz,1h),6.81(d,j=7.7hz,1h),4.65(d,j=3.8hz,1h),4.07(tt,j=6.7,3.2hz,1h),3.98(h,j=7.2hz,1h),3.88(p,j=7.3hz,1h),2.03(td,j=15.4,14.5,7.6hz,1h),1.77(ddt,j=12.4,7.9,4.2hz,1h),1.70–1.59(m,1h),1.62–1.42(m,3h),1.38(s,9h),1.38–1.29(m,1h),1.14(d,j=7.2hz,3h).
实施例9:式ivc化合物经过切酰胺键反应和boc保护反应得到式v化合物;
将10g式ivc化合物溶于60ml甲醇和20ml水中,室温下加入2.5g氢氧化锂,回流搅拌7h后,旋至不滴,加入20ml水,30ml二氯甲烷萃取三次。合并有机层,加入6.6gboc酸酐,室温搅拌30min。旋干反应液,柱层析得到4.6g油状物,收率65%。
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!