紫精衍生物、其制备方法与电致变色器件与流程
本发明涉及电致变色显示器领域,尤其涉及紫精衍生物、其制备方法与电致变色器件。
背景技术:
电致变色是材料的光学属性(反射率、透过率、吸收率等)在外加电场的作用下发生稳定、可逆的颜色变化的现象,在外观上表现为颜色和透明度的可逆变化。电致变色材料作为一种很有应用前景的新型功能材料,在大型显示、光电开关、电致变色存储器件、建筑窗玻璃及其灵巧窗等领域都有广泛的应用前景。
电致变色材料比需要具有良好的离子和电子导电性、较高的对比度、变色效率和循环周期性能。电致变色材料主要分为无机电致变色材料和有机电致变色材料。无机电致变色材料主要是以wo3等过度金属氧化物为代表,这些过渡金属氧化物通过离子和电子的共注入和共抽出,使其化学价态或晶体结构发生变化,从而实现着色和褪色的可逆过程;有机电致变色材料主要是以紫精等有共轭体系的分子为代表,是通过电子得失发生的氧化还原反应来实现着色和褪色的可逆变化,其包括有机小分子,如紫精;金属配位络合物,如酞化菁;有机导电聚合物,如聚苯胺、聚吡咯或聚噻吩等。
有机电致变色材料中的紫精(n,n-而取代-4,4-联吡啶)类材料自1932年发现以来在电致变色领域受到了广泛研究。紫精分子具有相当优秀的电致变色性能,基于紫精制备的电致变色器件在显示技术上有巨大的优势。但目前对紫精分子所做的研究仅限于n,n-二取代基的改变,这使得紫精材料难以满足多功能化的研究需求。
技术实现要素:
本发明解决的技术问题在于提供一种紫精衍生物,本申请提供的紫精衍生物使电致变色器件具有较好的电致变色性能。
有鉴于此,本申请提供了一种如式(ⅰ1)、式(ⅰ2)或式(ⅰ3)所示的紫精衍生物;
其中,r1、r2和r3独立的选自取代的c1~c5的烷基或未取代的c1~c5的烷基。
优选的,所述r1、r2和r3独立的选自未取代的c1~c5的烷基。
优选的,所述紫精衍生物如式(ⅰ1')、式(ⅰ2')或式(ⅰ3')所示;
本申请还提供了一种如式(ⅰ1)所示的紫精衍生物的制备方法,包括以下步骤:
a)将4-吡啶硼酸、具有式(ⅰ11)结构的化合物在催化剂的作用下反应,得到具有式(ⅰ12)结构的初始反应物;
b)将所述初始反应物与具有式(ⅰ13)结构的化合物在溶剂中反应,得到具有式(ⅰ14)结构的前驱体;
c)将所述前驱体与含有六氟磷酸根的化合物进行阴离子交换,得到如式(ⅰ1)所示的紫精衍生物;
其中,x1和x2独立的选自卤素;
r1选自取代的c1~c5的烷基或未取代的c1~c5的烷基。
本申请还提供了一种如式(ⅰ2)所示的紫精衍生物的制备方法,包括以下步骤:
a)将4-吡啶硼酸与具有式(ⅰ21)结构的化合物在催化剂的作用下反应,得到具有式(ⅰ22)结构的初始反应物;
b)将所述初始反应物与具有式(ⅰ23)结构的化合物在溶剂中反应,得到具有式(ⅰ24)结构的前驱体;
c)将所述前驱体与含有六氟磷酸根的化合物进行阴离子交换,得到如(ⅰ2)所示的紫精衍生物;
其中,x1和x2独立的选自卤素;
r2选自取代的c1~c5的烷基或未取代的c1~c5的烷基。
本申请还提供了一种如式(ⅰ3)所示的紫精衍生物的制备方法,包括以下步骤:
a)将4-吡啶硼酸与具有式(ⅰ31)结构的化合物在催化剂的作用下反应,得到具有式(ⅰ32)结构的初始反应物;
b)将所述初始反应物与具有式(ⅰ33)结构的化合物在溶剂中反应,得到具有式(ⅰ34)结构的前驱体;
c)将所述前驱体与含有六氟磷酸根的化合物进行阴离子交换,得到如(ⅰ3)所示的紫精衍生物;
其中,x1和x2独立的选自卤素;
r3选自取代的c1~c5的烷基或未取代的c1~c5的烷基。
优选的,所述催化剂为四三苯基膦钯,步骤a)所述反应在二氧化环与水的混合溶剂中进行;所述含有六氟磷酸根的化合物为六氟磷酸锂或六氟磷酸铵。
优选的,所述催化剂为三(二亚苄基亚酮)二钯,步骤a)所述反应在二氧六环和水的混合溶剂中进行;所述含有六氟磷酸根的化合物为六氟磷酸锂或六氟磷酸铵。
优选的,所述催化剂为四三苯基膦钯,步骤a)所述反应在n,n-二甲基甲酰胺和水的混合溶剂中进行;所述含有六氟磷酸根的化合物为六氟磷酸锂或六氟磷酸铵。
本申请还提供了一种电致变色器件,包括电解液,所述电解液中包括紫精衍生物和对电极材料,所述紫精衍生物为上述方案所述的紫精衍生物或上述方案所述的制备方法所制备的紫精衍生物。
本申请提供了一种紫精衍生物,其是利用偶联反应将苯环、萘环以或苯并噻二唑环引入至紫精分子中的两个吡啶环中间,增大了紫精分子的共轭程度,改变了紫精π电子骨架的电子轨道分布,增强了其荧光性能,使得该紫精衍生物制备的电致变色器件具有极高的光学调制率、多变的着色态颜色、超长的循环稳定性以及稳定的荧光发射现象。
附图说明
图1为实施例1制备的化合物的核磁共振氢谱图;
图2为实施例2制备的化合物的核磁共振氢谱图;
图3为实施例3制备的化合物的核磁共振氢谱图;
图4为本发明电致变色器件结构示意图;
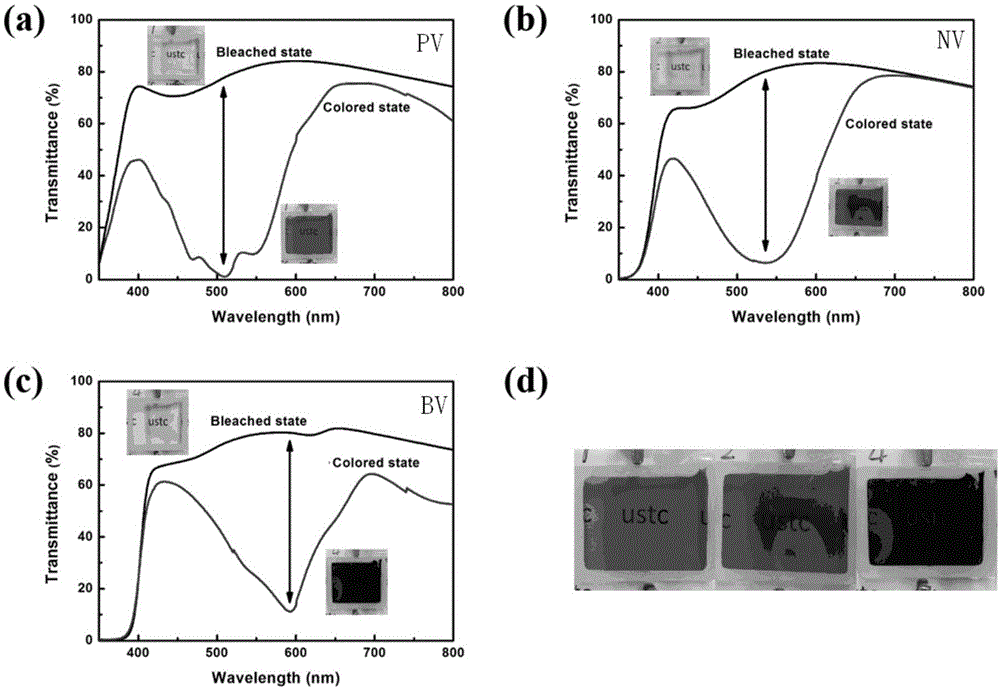
图5为本发明提供的紫精衍生物的电致变色性质曲线图;
图6为本发明提供的电致变色器件的荧光光谱与相应的荧光照片;
图7为本申请提供的电致变色器件的循环稳定性曲线图。
具体实施方式
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。
针对现在电致变色器件的现状,为了扩大紫精材料的多功能化,本发明实施例公开了紫精衍生物,本申请提供的紫精衍生物通过改变紫精的分子结构,得到了具有电致变色和荧光的双功能紫精衍生物,利用该材料制备的电致变色器件具有极高的光学调制率、多变的着色态颜色、超长的循环稳定性以及稳定的荧光发射现象;具体的,本申请所述紫精衍生物的结构如式(ⅰ1)、式(ⅰ2)或式(ⅰ3)所示:
其中,r1、r2和r3独立的选自取代的c1~c5的烷基或未取代的c1~c5的烷基。
更具体的,所述r1、r2和r3独立的选自未取代的c1~c5的烷基;更具体的,所述如式(ⅰ1)所示的紫精衍生物中,所述r1选自甲基;所述如式(ⅰ2)所示的紫精衍生物中,所述r2选自甲基;所述如式(ⅰ3)所示的紫精衍生物中,所述r2选自丁基;在上述情况下,所述紫精衍生物如式(ⅰ1')、式(ⅰ2')或式(ⅰ3')所示;其中如(ⅰ1')所示的紫精衍生物可简称为pv,如(ⅰ2')所示的紫精衍生物可简称为nv,如(ⅰ3')所示的紫精衍生物可简称为bv。
本发明还提供了所述紫精衍生物的制备方法,具体的:
所述如(ⅰ1)所示的紫精衍生物的制备方法,包括以下步骤:
a)将4-吡啶硼酸、具有式(ⅰ11)结构的化合物在催化剂的作用下反应,得到具有式(ⅰ12)结构的初始反应物;
b)将所述初始反应物与具有式(ⅰ13)结构的化合物在溶剂中反应,得到具有式(ⅰ14)结构的前驱体;
c)将所述前驱体与含有六氟磷酸根的化合物进行阴离子交换,得到如(ⅰ1)所示的紫精衍生物;
其中,x1和x2独立的选自卤素;
r1选自取代的c1~c5的烷基或未取代的c1~c5的烷基。
在上述制备如式(ⅰ1)所示的紫精衍生物中,首先将4-吡啶硼酸、具有式(ⅰ11)结构的化合物在催化剂的作用下反应,得到具有式(ⅰ12)结构的初始反应物;在制备上述初始反应物的过程中,具体是将4-吡啶硼酸、具有式(ⅰ11)结构的化合物、碳酸钾与催化剂在溶剂中混合反应,由此得到了初始反应物。在上述过程中,所述催化剂为四三苯基膦钯,所述溶剂为二氧六环和水的混合溶剂。
在得到初始反应物之后,将所述初始反应物与具有式(ⅰ13)结构的化合物在溶剂中反应,得到具有式(ⅰ14)结构的前驱体;所述溶剂优选采用三氯甲烷。
最后将上述得到的前驱体与含有六氟磷酸根的化合物进行阴离子交换,得到了具有式(ⅰ1)所示的紫精衍生物。在本申请中,所述含有六氟磷酸根的化合物选自六氟磷酸锂或六氟磷酸铵。
本申请还提供了如式(ⅰ2)所示的紫精衍生物的制备方法,包括以下步骤:
a)将4-吡啶硼酸与具有式(ⅰ21)结构的化合物在催化剂的作用下反应,得到具有式(ⅰ22)结构的初始反应物;
b)将所述初始反应物与具有式(ⅰ23)结构的化合物在溶剂中反应,得到具有式(ⅰ24)结构的前驱体;
c)将所述前驱体与含有六氟磷酸根的化合物进行阴离子交换,得到如(ⅰ2)所示的紫精衍生物;
其中,x1和x2独立的选自卤素;
r2选自取代的c1~c5的烷基或未取代的c1~c5的烷基。
在上述制备如式(ⅰ2)所述的紫精衍生物的过程中,同样分为初始反应物的制备-前驱体的制备-紫精衍生物的制备,在上述三个步骤中,前驱体的制备与紫精衍生物的制备过程与如式(ⅰ1)所述的紫精衍生物的制备过程相同,区别仅在于原料的选择上,因此在此处不进行赘述。
在制备初始反应物的制备过程中,将4-吡啶硼酸与具有式(ⅰ21)结构的化合物在催化剂的作用下反应,得到具有式(ⅰ22)结构的初始反应物;具体的,将4-吡啶硼酸、磷酸钾与具有式(ⅰ21)结构的化合物混合,再与溶解有三环己基膦与催化剂的有机溶剂混合,反应,得到初始反应物;上述有机溶剂为二氧六环与水,所述催化剂选自三(二亚苄基亚酮)二钯。
本申请还提供了一种如式(ⅰ3)所示的紫精衍生物的制备方法,包括以下步骤:
a)将4-吡啶硼酸与具有式(ⅰ31)结构的化合物在催化剂的作用下反应,得到具有式(ⅰ32)结构的初始反应物;
b)将所述初始反应物与具有式(ⅰ33)结构的化合物在溶剂中反应,得到具有式(ⅰ34)结构的前驱体;
c)将所述前驱体与含有六氟磷酸根的化合物进行阴离子交换,得到如(ⅰ3)所示的紫精衍生物;
其中,x1和x2独立的选自卤素;
r3选自取代的c1~c5的烷基或未取代的c1~c5的烷基。
在上述制备如式(ⅰ3)所述的紫精衍生物的过程中,同样分为初始反应物的制备-前驱体的制备-紫精衍生物的制备,在上述三个步骤中,前驱体的制备与紫精衍生物的制备过程与如式(ⅰ1)所述的紫精衍生物的制备过程相同,区别仅在于原料的选择上,因此在此处不进行赘述。
对于初始反应物的制备,本申请将4-吡啶硼酸与具有式(ⅰ31)结构的化合物在催化剂的作用下反应,得到具有式(ⅰ32)结构的初始反应物;在上述制备初始反应物的过程中,为了反应充分,则将具有式(ⅰ31)结构的化合物、4-吡啶硼酸、碳酸钾与催化剂在有机溶剂中混合反应,得到初始反应物。所述催化剂为四三苯基膦钯,所述有机溶剂为n,n-二甲基甲酰胺与水的混合溶剂。
本申请通过suzuki偶联反应将苯环、萘环以及苯并噻二唑环引入至紫精分子中的两个吡啶环中,增大了紫精分子的共轭程度,再以烷基修饰吡啶环上的氮原子,由此得到了一系列具有不同电致变色性能的紫精衍生物。
本发明还提供了所述紫精衍生物在电致变色器件中的应用,具体为,本发明还提供了一种电致变色器件,其包括电解液,所述电解液中包括紫精衍生物与二茂铁,所述紫精衍生物为上述方案所述的紫精衍生物。
上述电致变色器件的制备过程按照本领域技术人员熟知的方式进行,具体的,将紫精衍生物与对电极材料二茂铁溶解在电解液中灌注于两片基板中间的夹层里,随后以紫外胶封装整个器件,本申请所述的电致变色器件的结构如图4所示。本申请提供的电致变色器件的工作原理具体为:当外界电路接通时,紫精衍生物在电场的作用下向阴极移动,在阴极的氧化铟锡(ito)导电玻璃上得到电子还原为阳离子自由基的形态,该自由基显示较深的颜色,此时器件处于着色态;当外界电路断开时,处于着色态的紫精衍生物阳离子自由基分子自由扩散至溶液中并与对电极材料进行电荷交换而恢复原有无色透明状态,此时器件处于褪色态。
本发明首次将苯、萘以及苯并噻二唑等芳香环引入到经典的有机小分子紫精(n,n-二取代-4,4-联吡啶)中,并以这些材料为基础做成三明治结构的电致变色器件,得到了全新的电致变色-荧光双功能器件。这些新型双功能电致变色器件具有极其优秀的电致变色性能,基于苯环、萘环以及苯并噻二唑环的三种紫精衍生物器件分别在512nm,544nm以及594nm处具有最大光学调制率,分别能达到76.9%,74.1%以及69.0%,对应的器件着色态颜色分别是桃红色,紫红色以及蓝紫色,如图5所示;除电致变色性质之外,基于这些紫精衍生物材料的器件在紫外光源的激发下能发射出高强度的不同颜色的荧光,如图6所示,三个材料的荧光在紫外光激发照射下分别显示出蓝色,亮蓝色两种荧光颜色。与此同时,器件在相对应的最大光学透过率波长下的循环寿命可达到一千次,如图7所示。
为了进一步理解本发明,下面结合实施例对本发明提供的紫精衍生物、其制备方法与应用进行详细说明,本发明的保护范围不受以下实施例的限制。
以下实施例中的原料均为市售产品。
实施例14,4-(1,4-亚苯基)双(1-甲基吡啶基)六氟磷酸锂盐(pv)的合成
在150ml的圆底烧瓶中加入1,4-二碘苯(1.0g,3.0mmol)、4-吡啶硼酸(1.0g,8.1mmol)、碳酸钾(1.2g,8.6mmol)、催化剂四三苯基膦钯(0.28g,0.24mmol)以及磁力搅拌子,用氩气保护反应体系,再将二氧六环与水4:1的混合溶剂100ml注射入圆底烧瓶内,反应体系回流搅拌48h;
反应结束后将体系冷却至室温,加入去离子水,用三氯甲烷萃取有机相,无水硫酸钠干燥,之后过滤旋干溶剂三氯甲烷,得到黏稠状粗产物,再用柱色谱纯化粗产物,得到无色油状液体1;
将化合物1(1g,4.3mmol)以及过量的碘甲烷加入圆底烧瓶中,加入磁力搅拌子,以三氯甲烷为溶剂,搅拌24h,得到不溶物,将沉淀过滤出,用三氯甲烷洗涤数次,烘干,得到白色固体2;
将白色固体2溶解在高纯水中,加入过量六氟磷酸锂进行阴离子交换,搅拌得到沉淀,过滤沉淀得到最终产物化合物pv。图1为化合物pv的核磁共振氢谱图;1hnmr(300mhz,dmso-d6,ppm):δ9.094-9.077(d,4h),8.643-8.626(d,4h),8.342(s,4h),4.370(s,6h)。
上述反应过程如下式所示;
实施例24,4-(萘-1,4-二基)双(1-甲基吡啶基)六氟磷酸盐(nv)的合成
在150ml的圆底烧瓶中加入1,4-二溴萘(1.0g,3.5mmol)、4-吡啶硼酸(1.2g,9.7mmol)、磷酸钾(2.2g,10.3mmol)以及磁力搅拌子,用氩气保护反应体系,将三环己基膦(0.1g,0.35mmol)以及催化剂三(二亚苄基亚酮)二钯(0.16g,0.17mmol)溶解在二氧六环与水4:1的混合溶剂100ml中注射入圆底烧瓶内,反应体系回流搅拌24h;
反应的后处理、吡啶化合物的甲基化以及阴离子交换过程与实施例1的制备过程相同,最终得到化合物nv。图2为化合物nv的核磁共振氢谱图,1hnmr(300mhz,dmso-d6,ppm):δ9.261-9.239(d,4h),8.466-8.444(d,4h),8.028-7.994(m,2h),7.930(s,2h),7.855-7.824(m,2h),4.546(s,6h)。
上述反应过程如下式所示;
实施例34,4-(苯并[c][1,2,5]噻二唑-4,7-二基)双(1-丁基吡啶基)六氟磷酸盐(bv)的合成
在150ml的圆底烧瓶中加入4,7-二溴-2,1,3-苯并噻二唑(2.9g,10.0mmol)、4-吡啶硼酸(3.7g,30.0mmol)、碳酸钾(4.1g,30.0mmol)、催化剂四三苯基膦钯(0.50g,0.43mmol)以及磁力搅拌子,用氩气保护反应体系,再将n,n-二甲基甲酰胺与水3:1的混合溶剂100ml注射入圆底烧瓶内,反应体系回流搅拌24h;
反应的后处理、吡啶化合物的烷基化以及阴离子交换过程与实施例1的反应过程相同,最终得到化合物bv。图3为化合物bv的核磁共振氢谱图,1hnmr(300mhz,dmso-d6,ppm):δ9.305-9.288(d,4h),8.911-8.895(d,4h),8.608(s,2h),4.711-4.675(t,4h),2.020-1.983(m,4h),1.410-1.354(m,4h),0.990-0.953(t,6h)。
实施例4电致变色器件组装与性能测试
采用紫外固化胶将两片清洗过的氧化铟锡(ito)玻璃(3.45cm*3.45cm)相对固定,采用5ml注射器在两片氧化铟锡玻璃中间的空隙中注入溶解有紫精衍生物以及二茂铁的碳酸丙二醇酯/高氯酸锂(pc/liclo4)电解液,再将注射口用紫外胶封装固化,得到完整的电致变色器件。本申请电致变色器件的结构示意图如图4所示。
基于紫精衍生物(pv、nv、bv)的电致变色器件的电致变色性能测试通过联用chi660d电化学工作站与紫外-可见光-红外光谱仪,通过测量器件着色态与褪色态在300nm至800nm范围内的光学透过率得到,其结果如图5所示:三种电致变色器件在褪色态时均表现出高度的透明状态,光学透过率在400nm至800nm的范围内接近80%;在外加电场的作用下器件转变为着色态之后,三种器件着色态透过率最低值分别出现在512nm、544nm以及594nm处,透过率均在10%以下,所对应的最大光学透过率差值分别为76.9%、74.1%以及69.0%,如此优秀的光学调制率在电致变色领域也较为罕见。另外,三种器件在着色态对应的颜色如图5中所示,分别为桃红色,紫红色以及蓝紫色,相比于原本的紫精材料,这些衍生物的颜色种类更加丰富。
电致变色器件的荧光性质通过jobinyvonflurolog-3-tav荧光光谱仪测定,其结果如图6所示,器件在365nm紫外光源的激发下荧光发射峰分别出现在429nm,432nm以及463nm处,表现为蓝色以及亮蓝色的荧光,并且器件的荧光强度不因为电致变色过程中紫精衍生物的电化学还原过程而淬灭,也就是说电致变色器件在着色态以及褪色态都能维持高强度的荧光,这表示这些基于紫精衍生物的器件在黑暗环境的显示技术领域具有潜在的应用价值。
器件的循环稳定性测试为利用chi660d电化学工作站,对每个器件施加30s相对应的电压使其着色、100s的断路状态令其褪色,如此循环100、300、500以及1000次之后测量器件在300nm-800nm可见光范围内的光学透过率参数,不同次数间进行比较,直观地表征器件的循环稳定性。循环稳定性的结果如图7所示,三种基于不同紫精衍生物(pv、nv和bv)的器件在100、300、500以及1000次电致变色循环之后着色态与褪色态的透过率仅有微弱衰减,这表明器件具有优秀的循环稳定性,这为实现这些紫精衍生物在电致变色领域的应用打下了基础。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!