沉默荣昌猪CD14表达的siRNA及筛选方法与应用与流程
本发明属于生物基因工程及生物技术药物技术领域,具体涉及一种沉默荣昌猪cd14表达的小核糖核酸分子(sirna),本发明还涉及一种沉默荣昌猪cd14表达的sirna的筛选方法与应用。
背景技术
白细胞分化抗原14(clusterofdifferentiationantigen14,cd14)即为脂多糖(lipopolysaccharide,lps)高亲和性受体,借助于糖基磷脂酰肌醇(glycosylphosphatidylinosi-tol,gpi)锚固于单核细胞、巨噬细胞等髓样细胞膜的表面,cd14分为两种:膜结合型cd14(membranecd14,mcd14)和可溶性cd14(solublecd14,scd14)。
mcd14是一种分子量大小为55kd的膜蛋白,主要分布于单核细胞、巨噬细胞和树突细胞的表面,被激活的中性粒细胞表面也有少量的mcd14;scd14是一种糖蛋白,其分子量大小约为48kd,主要存在于正常人和动物的血浆中。
cd14主要功能是识别并结合lps,或与lps结合蛋白(lps-bindingprotein,lbp)形成复合物,激活toll样受体4(toll-likereceptor4,tlr4)信号通路,使之发生同源二聚化,然后通过两种途径激活靶细胞,实现lps信号的跨膜转导,在lps性炎症反应、内毒素休克等病理反应中起重要作用。因此,如果能够特异性地阻断cd14与lps及lps-lbp复合物的结合,就能够防治lps性炎症反应、内毒素休克等病理反应的发生,对于临床治疗内毒素血症、内毒素休克将会有非常重大的意义。
荣昌猪遗传资源丰富,肉质、鬃质和毛色特性优良,近年来开展了较为全面的遗传资源保护与开发利用工作。荣昌猪主要产于重庆市荣昌区和四川省的隆昌县而闻名,至今已有400多年的历史。在1986年,荣昌猪就被列入《中国猪品种志》,2003年农业部将其列入国家级保种名录,2006年农业部又将其列入《国家级畜禽遗传资源保护名录》。
针对荣昌猪cd14基因介导的信号通路和调控临床疾病分子机制的研究,几乎没有相关报道。猪cd14基因位于第二号染色体上,可编码373个氨基酸,liu等报道了猪cd14基因存在-61、587和1246等3个多态位点,并分析了-61位点的3种基因型与igg、dth等部分免疫性状之间的关系,结果发现:各免疫性状在不同基因型之间差异不显著。吴嘉韵等将梅山猪cd14基因-61位点的多态性与繁殖性能进行关联分析,也发现其不能作为繁殖性能的分子遗传标记。
核糖核酸干扰(rnainterference,rnai)是指由特定双链核糖核酸(doublestrandedrna,dsrna)引发同源信使核糖核酸(messengerrna,mrna)降解的转录后基因沉默机制。这类特定双链核糖核酸被裂解成小干扰核糖核酸分子(sirnas),并能导致同源信使核糖核酸在核糖核酸诱导的静默复合体(rna-inducedsilencingcomplex,risc)作用下降解。核糖核酸干扰rna是目前简单而高效地阻断哺乳动物细胞内特异基因表达的最有效方法之一,可达到基因敲除效果,且安全性好。但对于核糖核酸干扰来说,并不是所有针对靶基因信使核糖核酸序列随机设计的sirnas都能引起有效的基因沉默,干扰靶序列的选择对干扰效率的影响是至关重要的。因此,对能有效抑制特异基因表达的sirnas的筛选是应用核糖核酸干扰技术进行基因功能、基因治疗等相关研究的重要基础。
技术实现要素:
本发明的目的是提供一种沉默荣昌猪cd14表达的sirna,能够有效沉默荣昌猪cd14基因的表达,为开展靶向荣昌猪cd14的基因治疗荣昌猪内毒素休克、内毒素血症以及格兰氏阴性菌导致的炎症性疾病的研究提供了依据。
本发明的第二个目的是提供一种沉默荣昌猪cd14表达的sirna的筛选方法。
本发明的第三个目的是提供一种沉默荣昌猪cd14表达的sirna的应用。
本发明所采用的第一个技术方案是,一种沉默荣昌猪cd14表达的sirna,sirna的序列信息为:
正义,auugucagauagguccagggu(5′–3′);
反义,ccuggaccuaucugacaaucc(5′–3′)。
本发明所采用的第二个技术方案是,一种沉默荣昌猪cd14表达的sirna的筛选方法,包括以下步骤:
步骤1,构建重组表达质粒pcd14-egfp;
步骤2,构建稳定表达荣昌猪cd14基因的cd14-egfp稳定细胞系;
步骤3,根据cd14基因的序列,合成针对cd14基因的四个sirnas;
步骤4,使用高效转染试剂,将步骤3中四条sirnas转染步骤2的cd14-egfp稳定细胞系;
步骤5,经过步骤4的转染36h后,利用实时荧光定量聚合酶链式反应检测sirna的抑制效果,从而筛选出其中能特异且最有效沉默荣昌猪cd14基因表达的sirna。
本发明的特征还在于,
步骤1具体为:
步骤1.1,根据猪cd14基因序列,同时参照pegfp-n1载体信息,设计cd14真核表达引物f、r,并将pegfp-n1载体多克隆位点上的apai和ecori分别作为上、下游引物的酶切位点;
其中,cd14真核表达引物f、r具体为:
f:5’-gcgcgggcccatggtgagtgtgtgcacgagg-3’;
r:5’-cgcggaattcttaggcgaagccccggg-3’;
真核表达引物f、r中下划线位置处分别为pegfp-n1载体多克隆位点上的apai和ecori的上、下游引物的酶切位点;
步骤1.2,以cdna为模板,取步骤1.1中真核表达引物f、r进行降落pcr扩增cd14cds编码区,随后将扩增出的目的片段电泳,最后纯化回收扩增出的目的片段;
步骤1.3,将步骤1.1的pegfp-n1载体与步骤1.2的目的片段同时经apai和ecori双酶切消化,经电泳回收到线性化载体与扩增产物;然后在t4dna连接酶的介导下4℃过夜连接,将连接产物转化大肠杆菌dh5α感受态细胞,经iptg蓝白斑筛选,挑取单克隆进行菌落pcr初步鉴定,将阳性重组子摇菌,提取质粒,apai和ecori双酶切鉴定,测序得到重组表达质粒pcd14-egfp。
步骤1.2中扩增cd14cds编码区反应总体系50μl:模板cdna0.5μl,上、下游引物各1μl,2×taqpcrmastermix26μl,灭菌蒸馏水21.5μl;
降落pcr扩增条件为:95℃预变性2min,94℃变性20s、58℃退火20s,72℃延伸3min;依次循环以上扩增条件;当从第2个循环开始,每个循环降落1℃,直至退火温度降到50℃,再按照95℃预变性2min,94℃变性20s,50℃退火20s,72℃延伸3min进行30个循环反应;最后,72℃后延伸15min。
步骤2的具体步骤为:
步骤2.1,将猪肺泡巨噬细胞在含体积分数10%的fbs的杜比克改良伊格氏完全培养基中,于温度37℃、5%的co2的饱和湿度下培养,在转染的前一天,计数猪肺泡巨噬细胞并铺于6孔板中,确保转染时细胞密度达到80-90%;
步骤2.2,用250μl无血清的优化改良伊格氏完全培养基i分别稀释2.5μg步骤1重组表达质粒pcd14-egfp和8μl转染试剂,在室温孵育5min后,将两者轻微混匀后于室温孵育15min,得到脱氧核糖核酸-转染试剂复合物;
步骤2.3,将步骤2.2的脱氧核糖核酸-转染试剂复合物直接加入含细胞和无双抗培养基的6孔板中,混匀;待转染24h后,换完全培养基培养12h,同时使用0.25%胰蛋白酶,即含有0.02%edta的消化细胞,按1:4-9的比例稀释传代,待细胞贴壁后,在含有氨基糖苷类抗生素的选择性培养基中加压筛选,获得具有抗生素抗性的阳性细胞克隆;采用极限稀释法将克隆稀释至96孔板进行单克隆筛选,再将筛选出的单克隆扩增培养,从而获得cd14-egfp稳定细胞系。
步骤3中四个sirnas和使用的阴性对照具体如下所示:
步骤3中四个sirnas的合成过程中均遵循以下原则:
1)gc含量在30%-50%之间;
2)sense链的第15-19碱基中含有不少于3个a/u;
3)不含反向重复序列;
4)sense链的第19碱基为a;
5)sense链的第3碱基为a;
6)sense链的第10碱基为u;
7)sense链的第19碱基不是g/c;
8)sense链的第13碱基不是g。
步骤4的转染具体步骤为:
步骤4.1,在12孔板内放入200μl完全培养基,同时将8×105个步骤2的cd14-egfp稳定细胞系铺于12孔板内;
步骤4.2,用100μl无血清的opti-memi培养基稀释3μl浓度为20μm的sirnas,用100μl无血清的opti-memi培养基稀释8μl高效转染试剂转染试剂,然后将两个稀释物混合,充分搅拌均匀后形成转染复合物,在室温放置15min;
步骤4.3,将步骤4.2的的转染复合物滴入细胞培养孔内,轻摇混合均匀,然后放入37℃的二氧化碳培养箱中培养;在培养6h后,添加700μl杜比克改良伊格氏完全培养基,完成转染。
步骤5的实时荧光定量聚合酶链式反应中,合成靶向cd14基因的引物为:
上游引物为:5′-gcgctaacctcccctctctt-3′,
下游引物为:5′-gtcgtctatttggcagggct-3′。
本发明所采用的第三个技术方案是,一种沉默荣昌猪cd14表达的sirna作为治疗向荣昌猪cd14的基因介导的荣昌猪内毒素休克、内毒素血症以及格兰氏阴性菌引起的炎症性疾病的生物制药中的应用。
本发明的有益效果是:本发明筛选方法通过先建立稳定表达荣昌猪cd14基因的cd14-增强型绿色荧光蛋白egfp融合蛋白的猪肺泡巨噬细胞;然后合成四条位点不同的靶向cd14基因的sirnas,再应用高效转染试剂将四条sirnas,转染cd14-egfp稳定细胞系;最后筛选出能高效抑制荣昌猪cd14基因表达的sirna,即第3条(669)sirna能够明显抑制荣昌猪cd14基因的表达,为开展靶向荣昌猪cd14的基因治疗荣昌猪内毒素休克、内毒素血症以及格兰氏阴性菌导致的炎症性疾病的相关研究提供了的理论依据。
附图说明
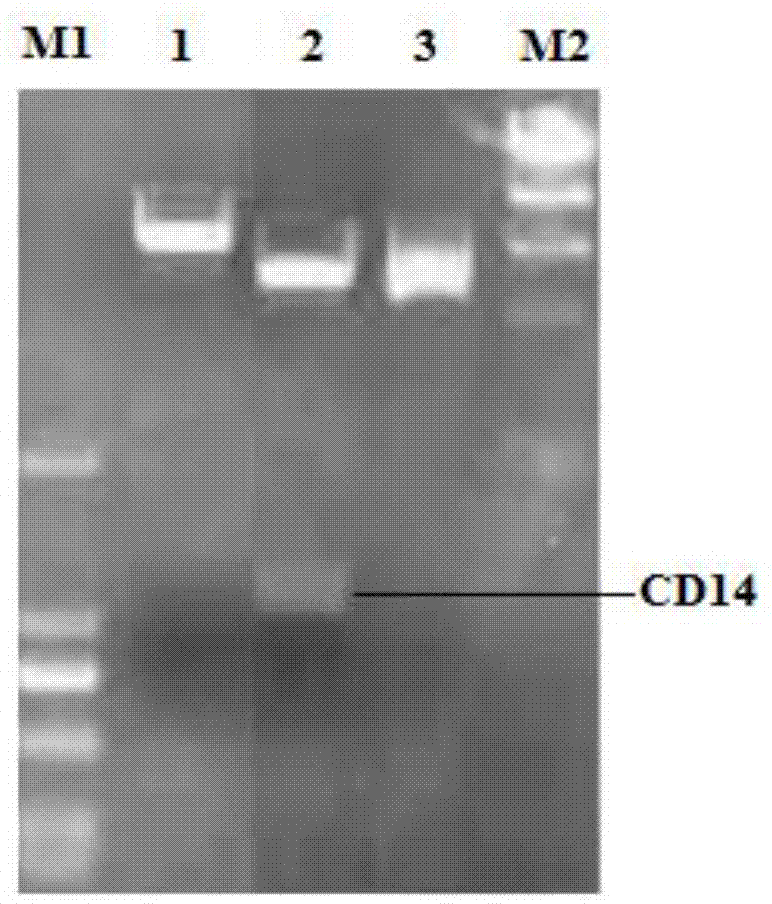
图1是本发明重组表达质粒pcd14-egfp的鉴定图;
图2是本发明稳定表达荣昌猪cd14基因的猪肺泡巨噬细胞的鉴定图,其中图2a为稳定表达空载egfp-n1的稳定细胞系和稳定表达cd14-egfp的稳定细胞系,图2b为稳定细胞系的western-blot鉴定结果;
图3是本发明利用实时荧光定量聚合酶链式反应检测sirna抑制cd14基因的mrna水平示意图。
具体实施方式
下面结合附图和具体实施方式对本发明进行详细说明。
本发明一种沉默荣昌猪cd14表达的sirna,首先通过先建立稳定表达荣昌猪cd14基因的cd14-增强型绿色荧光蛋白egfp融合蛋白的猪肺泡巨噬细胞(porcinealveolarmacrophagescells,pam);然后根据cd14基因的序列,设计合成四条不同的靶向cd14的sirnas、应用高效转染试剂将四条sirnas转染稳定表达的细胞系;最后利用实时荧光定量聚合酶链式反应鉴定其干扰效果,筛选出能高效抑制cd14基因表达的sirna。
综合上述的实验数据,发现第3条(669)sirna能够明显抑制cd14基因的表达。
具体操作步骤如下:
步骤1、重组表达质粒pcd14-egfp的构建
根据cd14基因序列(genbank登录号:ay753180.1),同时参照pegfp-n1载体信息,设计cd14真核表达引物:
f:5’-gcgcgggcccatggtgagtgtgtgcacgagg-3’;
r:5’-cgcggaattcttaggcgaagccccggg-3’;
将pegfp-n1载体多克隆位点上的apai和ecori分别作为上、下游引物的酶切位点,具体如下划线所示,设计的引物送北京六合华大基因(广州)科技股份有限公司合成。
以cdna为模板,对cd14真核表达引物降落pcr扩增cd14cds编码区:
总反应体系50μl:模板cdna0.5μl,上、下游引物各1μl,2×taqpcrmastermix26μl,灭菌蒸馏水21.5μl;
扩增条件:95℃预变性2min,94℃变性20s、58℃退火20s,72℃延伸3min,依次循环以上扩增条件;当从第2个循环开始,每个循环温度降落1℃,直至退火温度降到50℃;再按照95℃预变性2min,94℃变性20s、50℃退火20s,72℃延伸3min进行30个循环反应;最后,72℃后延伸15min。
将扩增出的目的片段经1%琼脂糖凝胶电泳,并利用dna纯化试剂盒进行胶回收扩增出的目的片段。
将dna纯化后的扩增产物与pegfp-n1质粒同时经apai和ecori双酶切消化,1%琼脂糖凝胶电泳,利用dna纯化试剂盒回收线性化载体与扩增产物,在t4dna连接酶的介导下4℃过夜连接,将连接产物转化大肠杆菌dh5α感受态细胞,经iptg蓝白斑筛选,挑取单克隆进行菌落pcr初步鉴定,将阳性重组子摇菌提取质粒,apai和ecori双酶切鉴定,送生工生物工程上海有限公司测序。
测序正确的重组质粒命名为:pcd14-egfp。
具体酶切鉴定结果如图1所示:m1为d2000dnamarkerλdna/hindⅲdnamarker;1为重组质粒pcd14-egfp经apai单酶切产物;2为重组质粒pcd14-egfp经apai和ecori双酶切产物;m2为λdna/hindⅲdnamarker,经分析表明,pcd14-egfp构建正确。
步骤2,构建稳定表达cd14基因的cd14-egfp稳定细胞系
将猪肺泡巨噬细胞在含体积分数10%的fbs的杜比克改良伊格氏完全培养基(dulbecco'smodifiedeagle'smedium,dmem)中,于温度37℃、5%的co2的饱和湿度下培养,按照转染试剂(lipofectaminetm3000,invitrogen,usa)操作说明书将重组质粒pcd14-egfp转染入猪肺泡巨噬细胞内,具体如下:
在转染的前一天,计数猪肺泡巨噬细胞并铺于6孔板中,确保转染时细胞密度达到80-90%;用250μl无血清的优化改良伊格氏完全培养基i分别稀释2.5μg步骤1的重组表达质粒pcd14-egfp和8μl转染试剂,在室温孵育6min后,将两者轻微混匀后于室温孵育15min,得到脱氧核糖核酸-转染试剂复合物;
将脱氧核糖核酸-转染试剂复合物直接加入含细胞和无双抗培养基的6孔板中,十字形轻摇以混匀;转染24h后,换完全培养基培养12h,同时使用0.25%胰蛋白酶(0.02%edta)消化细胞,按1:5的比例稀释传代,待细胞贴壁后,在含有氨基糖苷类抗生素(g418,500μg/ml)的选择性培养基中加压筛选,获得具有抗生素抗性的阳性细胞克隆;采用极限稀释法将克隆稀释至96孔板进行单克隆筛选,再将筛选出的单克隆扩增培养,从而获得稳定表达cd14-egfp融合蛋白的猪肺泡巨噬细胞。
对稳定表达cd14-egfp融合蛋白的猪肺泡巨噬细胞进行荧光显微镜检测和westernblot鉴定,结果如图2所示,图2a为稳定表达空载egfp-n1的稳定细胞系和稳定表达cd14-egfp的稳定细胞系,图2b为稳定细胞系的western-blot鉴定结果。
利用pbs洗涤细胞层1-2次,0.25%的胰酶(含有0.02%edta)常规消化,收集细胞。运用ip裂解液进行裂解,提取总蛋白,利用二喹林甲酸蛋白测定试剂盒测定总蛋白浓度,将总蛋白等量上样,在12%的分离胶的浓度下进行聚丙烯酰胺凝胶电泳(sds-page),随后进行蛋白质印迹(western-blot),主要的抗体:
一抗为兔抗cd14多克隆抗体(rabbitpolyclonalanti-cd14,1:1000倍稀释),兔抗甘油醛-3-磷酸脱氢酶单抗(gapdhrabbitmab,1:5000倍稀释);二抗为辣根过氧化物酶标记山羊抗兔免疫球蛋白g(igg)(hrplabeledgoatanti-rabbitigg,1:5000倍稀释)。
分析表明:稳定表达cd14-egfp融合蛋白的肺泡巨噬细胞正确表达荣昌猪cd14-增强型绿色荧光蛋白egfp融合蛋白,而对照则不能正确表达,说明稳定细胞系构建成功。
步骤3,根据cd14基因的序列,遵循sirna设计的基本原则,合成针对cd14基因的四个sirnas。
基本原则为:
1)gc含量在30%-50%之间;
2)sense链的第15-19碱基中含有不少于3个a/u;
3)不含反向重复序列;
4)sense链的第19碱基为a;
5)sense链的第3碱基为a;
6)sense链的第10碱基为u;
7)sense链的第19碱基不是g/c;
8)sense链的第13碱基不是g;
针对cd14基因的四个sirnas,如表1所示:
表1四个sirnas和阴性对照表
步骤4,使用高效转染试剂,将步骤3中四个sirnas分别转染步骤2的cd14-egfp稳定细胞系,具体为:
在12孔板内放入200μl完全培养基,同时将8×105个步骤2的cd14-egfp稳定细胞系铺于12孔板内;
用100μl无血清的opti-memi培养基稀释3μl浓度为20μm的sirnas,用100μl无血清的opti-memi培养基稀释8μl高效转染试剂转染试剂,然后将两个稀释物混合,充分搅拌均匀后形成转染复合物,在室温放置15min;
将转染复合物滴入细胞培养孔内,十字形轻摇以混合均匀,然后放入37℃的二氧化碳培养箱中培养;培养6h后,添加700μl杜比克改良伊格氏完全培养基,完成转染。
步骤5,经过步骤4的转染48h后,利用实时荧光定量聚合酶链式反应检测sirna的抑制效果,从而筛选出其中能特异且最有效沉默荣昌猪cd14基因表达的sirna。
(1)实时荧光定量聚合酶链式反应
转染48h后,吸出培养液,用磷酸盐缓冲液(pbs)洗涤细胞层1-2次,用0.25%的胰酶(0.02%edta)进行常规消化,收集细胞,根据核糖核酸提取试剂盒说明书分离出高品质的总核糖核酸;并测定纯化的核糖核酸浓度,采用溴化乙锭染色电泳鉴定其完整性;使用第一链试剂盒合成互补脱氧核糖核酸;采用abi-7500系统,使用荧光定量聚合酶链式反应试剂盒(
上游引物为:5′-gcgctaacctcccctctctt-3′,
下游引物为:5′-gtcgtctatttggcagggct-3′;
根据猪甘油醛-3-磷酸脱氢酶(gapdh)序列(genbank登录号:nm_001206359),设计并合成靶向猪gapdh的qpcr引物:
上游引物为:5′-agtgaacggatttggccgc-3′,
下游引物为:5′-tctcatggttcacgcccatc-3′;
结果如图3所示,图3中的横坐标代表稳定表达cd14-egfp的稳定细胞系、cd14-egfp稳定细胞系转染scramble-sirna、cd14-egfp稳定细胞系转染gfp-sirna、cd14-egfp稳定细胞系转染sirna1、cd14-egfp稳定细胞系转染sirna2、cd14-egfp稳定细胞系转染sirna3、cd14-egfp稳定细胞系转染sirna4,纵坐标代表荧光强度的相对值,scramble-sirna是相对cd14无关的非特异sirnas。
结果表明:实时荧光定第3条sirna3(669)能靶向抑制cd14mrna水平,抑制效率约为70%。
以上信息为后续研究靶向荣昌猪cd14的基因治疗荣昌猪内毒素休克、内毒素血症以及格兰氏阴性菌导致的炎症性疾病和进一步应用核糖核酸干扰(rnai)技术通过抑制cd14基因的表达奠定了重要基础。
- 还没有人留言评论。精彩留言会获得点赞!