一种渥曼青霉素前药及其制备与应用的制作方法
本发明涉及一种渥曼青霉素前药及制备方法与应用。
(二)背景技术
产生自丝状真菌penicilliumfuniculosum的渥曼青霉素(wortmannin),对磷脂酰肌醇3-激酶pi3k具有很高的抑制活性,其半抑制浓度(ic50)大约为5nm,比常规抑制剂ly294002高了将近200倍。实验证明,其抑制机制为渥曼青霉素结构中的第20位的碳原子(c20位点)与pi3k催化活性中心的赖氨酸(lys833)侧链氨基相结合,形成烯胺。与此同时,烯胺中的氢与相邻的羰基形成氢键,生成了稳定的六元环结构,造成该位点的不可逆修饰(如图1所示),从而抑制pi3k及pi3k/akt信号传导通路,最终促进了肿瘤细胞的死亡(ic50在10μm左右)。然而,因渥曼青霉素的c20位点具有较高的反应活性,易受水及其它生物大分子中的亲核基团进攻,导致它在水溶液中的稳定性降低,毒性增强,故而限制了渥曼青霉素进一步的开发利用。
寻求并改善抗肿瘤药物,使其选择性富集在肿瘤部位,以此实现抗癌药物的靶向性,不仅能够提高治疗效果,还对减轻患者的痛苦、提高患者的生存质量等方面具有十分重要的现实意义。肿瘤细胞的细胞膜上会特异性表达或过量表达一系列受体或抗原,通过肿瘤靶向性传递系统(targeteddrugdeliverysystem)来满足肿瘤细胞增殖所需的各种营养,生物素(维他命b7,biotin)及其受体就是其中一种传递系统。
(三)
技术实现要素:
本发明目的是针对渥曼青霉素现有的不足,结合它c20位点的特性以及生物素的肿瘤靶向性,制备了多种含仲胺基团的渥曼青霉素前药。本发明提供的前药具有不同的缓释速率。同时,本发明提供的前药具有良好的抗肿瘤细胞增值及抑制pi3k/akt信号通路的性能。
本发明采用的技术方案是:
本发明提供一种式(ⅱ)所示渥曼青霉素前药,
式(ii)中,r1和r2各自独立为:带有生物素的四氢吡咯环、带有生物素的哌啶环、异丙基或带有生物素的二乙胺基。
进一步,所述式(ⅱ)所示渥曼青霉素前药为下列之一:
本发明还提供一种所述渥曼青霉素前药的制备方法,所述方法为:将式(i)所示化合物、三乙胺溶于二氯甲烷中,随后加入仲胺化合物,室温搅拌反应10小时,反应液经薄层色谱分离纯化后得到式(ⅱ)所示渥曼青霉素前药;所述仲胺化合物为式(iii)、式(iv)或式(v)所示化合物;
进一步,所述式(i)所示化合物与三乙胺物质的量之比为1:2-5(优选1:4.3),所述式(i)所示化合物与仲胺基团质的量之比为1:1-3(优选1:1.2);所述二氯甲烷体积用量以式(i)所示化合物重量计为1l/g。
进一步,所述分离纯化方法为:反应结束后,反应液通过减压旋蒸除去溶剂后直接利用高效加厚制备薄层板分离,展开剂为体积比10~20:1:0.1的二氯甲烷:甲醇:三乙胺,收集rf值0.2~0.6的样品,得到目标化合物。
本发明还提供一种所述渥曼青霉素前药在制备肿瘤细胞活性抑制剂中的应用。
进一步,所述肿瘤细胞为人乳腺癌细胞mcf-7、人肺癌细胞a549、人肝癌细胞hepg2或人宫颈癌细胞hela。
根据渥曼青霉素的结构特点及抑制机理,本发明利用二级胺(仲胺)对c20位点进行修饰,形成式ii。在生理条件下,分子内c6位点上的羟基可进攻c20位点,使仲胺基团缓慢离去,再次形成呋喃环,进而释放渥曼青霉素,以达到缓释的效果,如图3所示。
与现有技术相比,本发明有益效果体现在:本发明以渥曼青霉素为母体,利用简单的化学合成手段,构建了具有不同缓释效应的抑制剂。通过对体外缓释实验、癌细胞增值实验、akt磷酸化检测验证了,本发明的抑制剂仍保持着对pi3k通路影响效果的同时,利用了药物缓慢释放作及生物素靶向作用,提高了抗肿瘤靶向性,大大降低了抑制癌细胞增值的用量。
(四)附图说明
图1为渥曼青霉素对磷脂酰肌醇3-激酶pi3k的抑制机制。
图2为渥曼青霉素前药结构示意图(以化合物(ⅱ-1)为例)。
图3为本发明渥曼青霉素前药作用机制示意图。
图4为实施例1制备的化合物(ⅱ-1)的核磁氢谱。
图5为实施例1制备的化合物(ⅱ-2)的核磁氢谱。
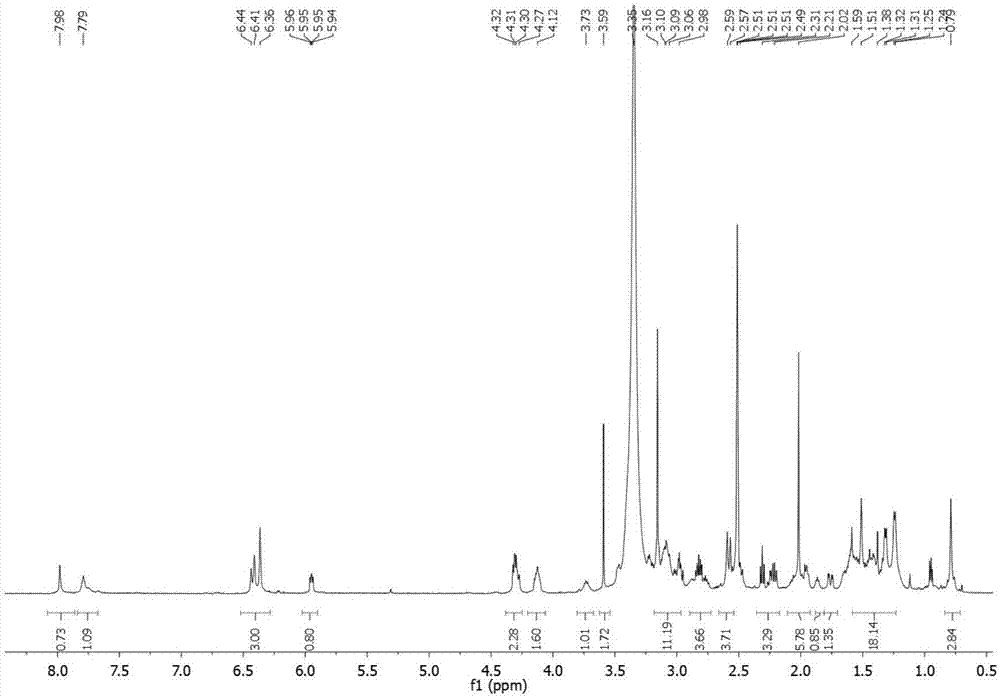
图6为实施例3制备的化合物(ⅱ-3)的核磁氢谱。
图7为渥曼青霉素前药与苯甲胺反应的产率曲线图。
图8为渥曼青霉素前药(ⅱ-1)与苯甲胺反应流程示意图。
图9为渥曼青霉素(i)和化合物(ⅱ-1)、(ⅱ-2)、(ⅱ-3)在人乳腺癌细胞mcf-7中的抗肿瘤增殖效果图。
图10为渥曼青霉素(i)和化合物(ⅱ-1)、(ⅱ-2)、(ⅱ-3)在人肺癌细胞a549中的抗肿瘤增殖效果图。
图11为渥曼青霉素(i)和化合物(ⅱ-1)、(ⅱ-2)、(ⅱ-3)在人宫颈癌细胞hela中的抗肿瘤增殖效果图。
图12为渥曼青霉素(i)和化合物(ⅱ-1)、(ⅱ-2)、(ⅱ-3)在人肝癌细胞hepg2中的抗肿瘤增殖效果图。
图13为渥曼青霉素(i)和化合物(ⅱ-1)、(ⅱ-2)、(ⅱ-3)对pi3k/akt通道影响的免疫印迹图。
(五)具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
本发明实施例所述室温是指25-30℃。
实施例1化合物(ii-1)的制备
在25ml的圆底烧瓶中,将化合物i(10mg,23μmol)与三乙胺(16μl,100μmol)溶于10ml二氯甲烷后,加入化合物iii(9mg,28μmol),室温搅拌反应10小时后,真空抽干溶剂,利用高效加厚制备薄层板分离,展开剂为体积比20:1:0.1的二氯甲烷:甲醇:三乙胺,收集rf值为0.6的样品,得到目标化合物(ii-1)(7mg,43%),核磁氢谱见图4。
1hnmr(500mhz,dmso)δ8.69(s,1h),8.02(s,1h),6.42(s,1h),6.37(s,1h),5.95(dd,j=7.1,4.6hz,1h),4.31(t,j=6.5hz,2h),4.14(s,1h),3.64-3.73(m,3h),3.04-3.22(m,8h),2.90-3.04(m,3h),2.75-2.84(ddd,j=19.5,11.7,5.5hz,3h),2.53-2.63(m,3h),2.20-2.38(m,5h),1.99-2.09(m,4h),1.76(d,j=13.9hz,1h),1.42-1.68(m,8h),1.27-1.40(s,2h),0.78(s,3h).hrms(esi):m/z[m+na]+calcd763.2989,found:763.3017.
实施例2化合物(ⅱ-2)的制备
在25ml的圆底烧瓶中,将化合物i(10mg,23μmol)与三乙胺(16μl,100μmol)溶于10ml二氯甲烷后,加入化合物iv(11mg,28μmol),室温搅拌反应10小时后,真空抽干溶剂,利用高效加厚制备薄层板分离,展开剂为体积比12:1:0.1的二氯甲烷:甲醇:三乙胺,收集rf值为0.5的样品,得到目标产物(ii-2)(10mg,52%),核磁氢谱见图5。
1hnmr(500mhz,dmso)δ8.48(s,1h),7.81(s,1h),6.44(s,1h),6.36(s,1h),5.95(t,j=5.66hz,1h),5.04(t,j=8.92hz,1h),4.37(d,j=5.5hz,1h),4.27-4.34(m,1h),4.08-4.19(m,1h),3.44-3.66(m,7h),3.04-3.20(m,7h),2.91-3.09(m,4h),2.74-2.87(m,3h),2.66(s,1h),2.52-2.62(m,2h),2.42-2.46(m,1h),2.34(s,2h),2.12-2.27(m,4h),1.86-2.08(m,7h),1.70-1.78(m,2h),1.57-1.65(m,2h),1.42-1.54(m,7h),1.29-1.41(m,3h),0.75(s,4h).hrms(esi):m/z[m+na]+calcd860.3516,found:860.3534.
实施例3化合物(ⅱ-3)的制备
在25ml的圆底烧瓶中,将化合物i(10mg,23μmol)与三乙胺(16μl,100μmol)溶于10ml二氯甲烷后,加入化合物v(9mg,28μmol),室温搅拌反应10小时后,真空抽干溶剂,利用高效加厚制备薄层板分离,展开剂为体积比10:1:0.1的二氯甲烷:甲醇:三乙胺,收集rf值为0.23的样品,得到目标产物(ii-3)(9mg,46%),核磁氢谱见图6。
1hnmr(500mhz,dmso)δ7.98(s,1h),7.79(s,1h),6.32-6.47(m,3h),5.95(dd,j=7.3,4.5hz,1h),4.30(dd,j=15.4,9.6hz,2h),4.12(s,2h),3.73(s,1h),3.59(s,2h),2.94-3.19(m,11h),2.71-2.89(m,3h),2.53-2.63(m,5h),1.93-2.18(m,6h),1.87(s,1h),1.76(dd,j=14.3,4.0hz,1h),1.18-1.58(m,18h),0.78(s,3h).hrms(esi):m/z[m+na]+calcd779.3302,found:779.3310.
实施例4化合物(ⅱ-1)-(ⅱ-3)体外释放试验
分别将实施例1、2、3中制备的化合物(ⅱ-1)、(ⅱ-2)、(ⅱ-3)溶于1ml溶剂(水:乙腈:二甲基亚砜=2:2:2,v/v/v)中制成1mm前药溶液,取1ml前药溶液加入500μl、10mm苯甲胺溶液(溶剂为水:乙腈=1:1,v/v),依次在不同时间(0,1,2,4,6,8,10,13,23,43,57,81,107小时)进样5μl,获得高效液相色谱图。色谱条件:采用岛津hplc系统及phenomenexc18色谱柱,流动相系统为水和乙腈的比例和体积流量的梯度洗脱程序(流速0.6ml/min,5%-100%乙腈10分钟,5%乙腈5分钟),柱温35℃,检测波长254nm。利用色谱图中原料峰及产物峰的比例计算每个时间点的产率,并作产率与时间关系图,结果见图7所示。
从结果明显发现,因仲胺修饰的结构,分子内c6位点上的羟基可进攻c20位点可逆形成渥曼青霉素,紧接着快速与苯甲胺上的氨基反应生成稳定的产物vi(图8所示),因此,c20位点上的仲胺修饰直接导致了羟基进攻的速率。从图7结果表明,六元环修饰前药(ⅱ-1)的半衰期约10小时,五元环修饰前药(ⅱ-2)的半衰期约50小时,异丙基的修饰前药(ⅱ-3)的半衰期约136小时。因此三种修饰能达到不同程度的缓释效果。
实施例5抗肿瘤细胞增殖研究
本发明所述前药针对人肺癌细胞a549、人宫颈癌细胞hela、人肝癌细胞hepg2、人乳腺癌细胞mcf-7的细胞增值影响评价采用一种标准的mtt(3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐)方法。
将上述肿瘤细胞分别以悬液(40000/ml,dmem培养基)接种于96孔板,加样量为100μl/孔。以渥曼青霉素(i)、实施例1、2、3中制备的化合物(ⅱ-1)、(ⅱ-2)、(ⅱ-3)为实验对象,实验设置8个浓度梯度给药组(药物最终浓度分别为40μm,20μm,10μm,5μm,2.5μm,1.25,625nm,312.5nm,每个浓度的培养孔中添加体积终浓度1%dmso),1个不加药对照组(1μldmso),每组3个平行。将96孔板置于37℃,5%co2培养箱与药物孵育72个小时。孵育完毕后,加入10μlmtt(5mg/ml),继续孵育4个小时。随后吸出培养液,每孔加入150μldmso,将96孔板置于振荡器摇晃至紫色沉淀充分溶解,然后使用酶标仪测量550nm处的吸光度。利用对照孔计算增值率,并最后不同浓度下拟合计算得到母体渥曼青霉素(i)和本发明实施例1、2、3中制备的化合物的ic50值,重复三次试验得到标准偏差,如图8-12和表1所示。表1为渥曼青霉素(i)和化合物(ⅱ-1)、(ⅱ-2)、(ⅱ-3)的体外抗肿瘤细胞增殖研究结果
试验结果表明,通过对渥曼青霉素c20位点的优化修饰后,使得能在细胞内渥曼青霉素缓慢释放,延长了药物作用时间。修饰后的渥曼青霉素ic50值比其母体化合物的ic50值降低,说明经修饰后,使用更低的用药量即可达到肿瘤细胞的半抑制效果。
实施例6本发明所述靶向抗肿瘤药物对pi3k/akt通道的影响研究
渥曼青霉素一直被用作pi3k/akt信号通路的抑制剂,因此我们利用检测磷酸化akt的水平来验证这些前药对pi3k/akt信号的影响。
将a549细胞以20,000细胞/孔接种在含有500μldmem培养基的24孔板中,并置于37℃,5%co2培养箱12小时使得细胞贴壁。随后每个孔加入最终浓度为125μm的渥曼青霉素(i)以及实施例1、2、3中制备的化合物(ⅱ-1)、(ⅱ-2)、(ⅱ-3),随后置于37℃,5%co2培养箱孵育。在孵育不同时间(1,4,8,20小时)后,抽吸培养基,细胞用冰冷的pbs洗涤,将细胞刮下直接用1×sds上样缓冲液裂解得到蛋白。在15孔sds-page12%凝胶中每孔加入50μg总蛋白量。电泳后将蛋白转移到0.45μmpvdf膜上,用含3%bsa的tbst封闭一小时。抗体按比例用含3%bsa的tbst稀释,akt(cellsignaling,1:1000,孵育1小时),pakt(cellsingaling,1:1000,孵育1小时),山羊-抗-兔-igg-偶联-hrp(cellsingaling,1:3000,1小时)。适用supersignalwestdura化学发光底物(pierce)检测蛋白。结果如图13所示。从结果发现,这四种化合物都会对pi3k/akt通路有抑制作用。渥曼青霉素(i)以及化合物(ⅱ-1)和(ⅱ-2)可以相对较快地降低akt磷酸化程度,而化合物(ⅱ-3)的缓释效果使得抑制效果相对更持久。而化合物(ⅱ-3),因其可逆过程相对慢,虽然磷酸化的抑制效果没有其他的快速,但是从8小时到20小时的结果中发现化合物(ⅱ-3)的抑制效果更持久。
- 还没有人留言评论。精彩留言会获得点赞!