一种具有抑制组蛋白甲基化酶DOT1L二甲基化活性的多肽、抑制剂及其设计方法与应用与流程
本发明涉及生物医药工程技术领域,具体涉及一种具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽、抑制剂及其设计方法与应用。
背景技术
表观遗传学是20世纪80年代逐渐兴起的一门学科,是在研究与经典孟德尔遗传学遗传法则不相符的许多生命现象过程中逐步发展起来的。表观遗传学主要研究内容大致包括两个方面:1)基因选择性转录表达的调控,具体包括dna甲基化、基因印记、组蛋白共价修饰和染色质重塑等;2)基因转录后的调控,包括基因组中非编码的rna、微小rna、反义rna、内含子及核糖开关等。染色质的基本单位是核小体,核小体由dna与组蛋白构成。染色质的结构状态将影响基因表达,当组蛋白将dna围绕成压缩状态时,dna将不能转录;当组蛋白与dna形成疏松状态时,dna才能转录,因此,组蛋白和dna进行表观修饰可改变染色质结构状态从而导致基因激活或沉默。
组蛋白有五种类型,具体为h1、h2a、h2b、h3和h4,其中,组蛋白h3的氨基末端露在外面,可以进行共价修饰,如乙酰化与去乙酰化、磷酸化与去磷酸化、甲基化与去甲基化等。组蛋白甲基化是一个可逆过程,是由组蛋白甲基转移酶(histonemethyltransferase,hmt)和组蛋白去甲基化酶(histonedemethylase)共同参与形成和维持不同的组蛋白甲基化状态。
组蛋白甲基转移酶dot1l是近期新发现的一种甲基转移酶,在分子伴侣存在下,该酶通过与rna解螺旋酶和rna聚合酶相互作用,与一系列下游基因启动子区域的序列结合,进而影响靶基因的表达。在已知的组蛋白甲基化酶中,dot1l是唯一能催化组蛋白h3第79位赖氨酸单甲基化、二甲基化和三甲基化三种酶活反应的甲基化酶。dot1l可与不同的调节因子形成不同类型的大复合物,这些大复合物在很多的细胞活动中都发挥着各自不同的作用,其中包括癌基因转录激活、异染色质区域形成和促进转录延伸过程等。
af10蛋白主要通过结合并调节组蛋白h3k79位点甲基化酶dot1l复合物的二甲基化活性来完成的,它能调节dot1l二甲基化酶活性,是急性骨髓单核细胞性白血病中细胞增殖能力的关键因子,可作为抗白血病药物开发的新治疗靶点。美国纪念斯隆凯特琳癌症研究中心的研究人员发现af10蛋白可调控组蛋白甲基化酶dot1l的二甲基化活性,同时对急性骨髓单核细胞性白血病细胞的增殖有关键作用。此外,哥伦比亚大学的研究人员发现,af10蛋白能识别组蛋白上的表观遗传学修饰,并招募组蛋白甲基化酶到基因的启动子位置来实现基因的转录调控。
针对af10蛋白的药物设计是一个比较新颖的研究方向,若能成功,其将会特异性地抑制dot1l二甲基化酶活性,从而调节急性骨髓性白血病细胞中癌基因表达。同时,联合使用针对af10蛋白的药物与dot1l小分子抑制剂,还将优化dot1l酶活中心抑制小分子药物在白血病治疗中的应用效果。
目前,基于蛋白质结构信息而设计的dot1l酶活中心抑制小分子药物epz-5676在美国已进入临床二期实验,但是由于该小分子并不能区分抑制dot1l的单甲基化、二甲基化和三甲基化三种酶活反应,故急需对dot1l各种酶活调节因子的结构特征进行深入研究,并找出一种能够特异性抑制该酶二甲基化活性的物质。
参考文献:
1.chen,c.w.&armstrong,s.a.targetingdot1landhoxgeneexpressioninmllrearrangedleukemiaandbeyond.exphematol43,673-684,(2015).
2.nguyen,a.t.&zhang,y.thediversefunctionsofdot1andh3k79methylation.genesdev25,1345-1358,(2011).
3.vanleeuwen,f.,gafken,p.r.&gottschling,d.e.dot1pmodulatessilencinginyeastbymethylationofthenucleosomecore.cell109,745-756,(2002).
4.feng,q.etal.methylationofh3-lysine79ismediatedbyanewfamilyofhmtaseswithoutasetdomain.currbiol12,1052-1058,(2002).
5.min,j.,feng,q.,li,z.,zhang,y.&xu,r.m.structureofthecatalyticdomainofhumandot1l,anon-setdomainnucleosomalhistonemethyltransferase.cell112,711-723,(2003).
6.chen,s.etal.thepzpdomainofaf10sensesunmodifiedh3k27toregulatedot1lmediatedmethylationofh3k79.molcell60,319-327,(2015).
7.deshpande,a.j.etal.af10regulatesprogressiveh3k79methylationandhoxgeneexpressionindiverseamlsubtypes.cancercell26,896-908,(2014).
8.cecere,g.,hoersch,s.,jensen,m.b.,dixit,s.&grishok,a.thezfp-1(af10)/dot-1complexopposesh2bubiquitinationtoreducepoliitranscription.molcell50,894-907,(2013).
9.avgousti,d.c.,cecere,g.&grishok,a.theconservedphd1-phd2domainofzfp-1/af10isadiscretefunctionalmoduleessentialforviabilityincaenorhabditiselegans.molcellbiol33,999-1015,(2013).
10.daigle,s.r.etal.potentinhibitionofdot1lastreatmentofmll-fusionleukemia.blood122,1017-1025,(2013).
技术实现要素:
本发明所要解决的技术问题是:提供一种具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽、抑制剂及其设计方法与应用。
一种具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽,所述多肽的氨基酸序列如seqno.1所示。
进一步地,所述多肽的氨基酸序列为qiewakarveklrkrnqalksqtselqrqiaeleasnaelkk。
本发明的有益效果在于:本发明方案的多肽在体外能特异性结合甲基化酶活性调节因子af10,结合常数ka达到14nm。结构生物学研究结果表明,该多肽前体对af10的结合阻断了af10与组蛋白甲基化酶dot1l二甲基化活性的能力,从而阻断了af10分子参与调节组蛋白甲基化酶dot1l二甲基化活性的能力,最终实现对该酶活性的抑制作用。
本发明还包括一种抑制剂,所述抑制剂包括上述多肽。
进一步地,所述抑制剂还包括任选的一种或多种药学上可接受的赋形剂、稀释剂或载体。
本发明还包括一种具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽的设计方法,包括以下步骤:
s1、获取af10-dot1l复合物晶体结构,利用体内蛋白互补实验、噬菌体展示及酵母双杂交的实验系统,找出具有抑制dot1l结合的多肽前体;
s2、通过所述多肽前体与af10的复合物进行结晶学实验,获得af10-多肽前体复合物晶体;
s3、改造所述多肽前体序列,设计出具有与af10特异性结合的多肽片段;
s4、在s3的基础上进一步优化出具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽。
进一步地,所述步骤s1中获取af10-dot1l复合物晶体结构的操作为:鉴定af10与dot1l相互作用的区段后,通过共纯化或共表达的方式获得af10-dot1l相互作用区段的蛋白质复合物。
进一步地,所述鉴定af10与dot1l相互作用的区段的具体操作为通过二级结构预测法,利用分子克隆或大肠杆菌原核表达系统等分子生物学技术手段,共表达或分开表达出所述af10-dot1l复合物感兴趣的相互作用区段。
进一步地,所述步骤s1中,所述找出具有抑制dot1l结合的多肽前体的具体操作为:建立体内蛋白互补实验、噬菌体展示以及酵母双杂交的实验系统,鉴定与af10特异结合的多肽分子。
进一步地,所述步骤s2的具体操作为:通过结晶学实验方法,对af10-抑制性多肽分子复合物的晶体生长条件进行筛选和优化,并获得能够解析该晶体结构的衍射数据。
进一步地,所述步骤s3的具体操作为:利用af10-dot1l复合物晶体结构中参与相互作用的关键氨基酸信息,改造蛋白序列,从而设计出与af10特异性结合的多肽片段。
进一步地,所述步骤s4的具体操作为:在所述步骤s3的基础上进一步通过随机突变的方式对参与af10-dot1l相互作用的关键氨基酸进行优化,优化出能与dot1l蛋白分子竞争结合af10的抑制性多肽片段。
优选地,所述具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽的氨基酸序列如seqno.1所示。
本发明的有益效果在于:本发明通过结晶学实验研究dot1l酶活调节因子的结构特征进而筛选出能够特异性抑制组蛋白甲基化酶dot1l二甲基化活性的多肽前体,进而设计出能与af10特异性结合的多肽片段,科学合理且设计得到的多肽片段特异性强。
本发明还包括上述多肽在制备组蛋白去甲基化酶dot1l抑制剂药物中的应用。
进一步地,所述组蛋白去甲基化酶dot1l抑制剂药物为抗癌药物。
进一步地,所述抗癌药物为抗血癌药物。
从上述描述可知,本发明的有益效果在于:由于组蛋白甲基化酶dot1l二甲基化活性对白血病的发生发展起着至关重要的作用,而本发明抑制剂对于该酶具有良好的抑制作用,因此,本发明方案的抑制剂在抗癌药物中具有良好的应用前景,尤其是血癌(白血病)的治疗药物。
附图说明
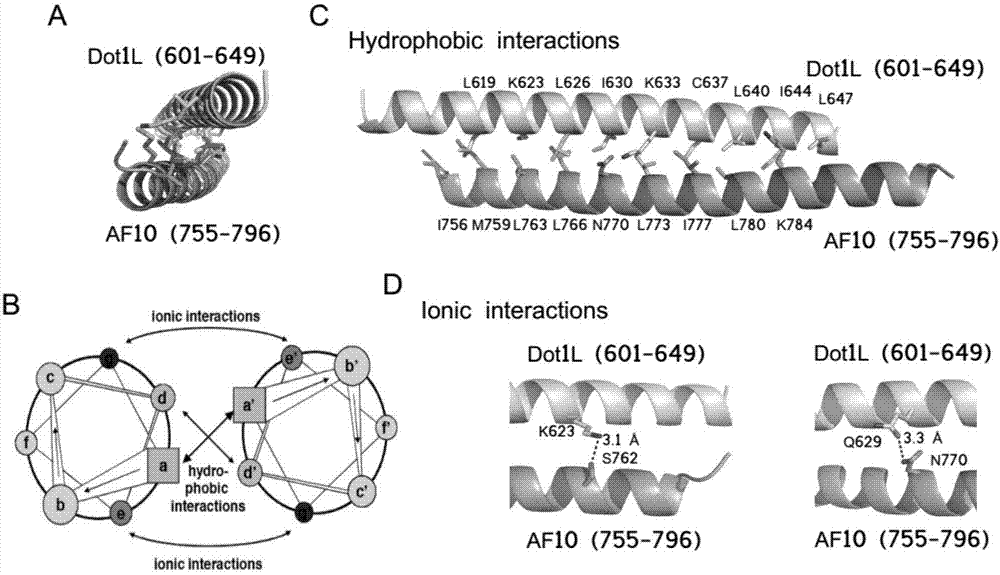
图1为本发明实施例的af10-dot1l复合物晶体空间结构示意图;
图2为本发明实施例多肽分子与af10复合物的结合晶体空间结构示意图;
图3为本发明实施例的af10分子与多肽分子的结合力曲线图;
图4为本发明实施例的多肽分子阻断af10分子参与调节组蛋白甲基化酶dot1l二甲基化活性的机理示意图。
具体实施方式
为详细说明本发明的技术内容、构造特征、所实现目的及效果,以下结合实施方式并配合附图详予说明。
一种具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽,所述多肽的氨基酸序列如seqno.1所示。
从上述描述可知,本发明的有益效果在于:本发明方案的多肽在体外能特异性结合甲基化酶活性调节因子af10,结合常数ka达到14nm。结构生物学研究结果表明,该多肽前体对af10的结合阻断了af10与组蛋白甲基化酶dot1l二甲基化活性的能力,从而阻断了af10分子参与调节组蛋白甲基化酶dot1l二甲基化活性的能力,最终实现对该酶活性的抑制作用。
进一步地,所述多肽的氨基酸序列为qiewakarveklrkrnqalksqtselqrqiaeleasnaelkk。
本发明还包括一种抑制剂,所述抑制剂包括上述多肽。
进一步地,所述抑制剂还包括任选的一种或多种药学上可接受的赋形剂、稀释剂或载体。
本发明还包括一种具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽的设计方法,包括以下步骤:
s1、获取af10-dot1l复合物晶体结构,利用体内蛋白互补实验、噬菌体展示及酵母双杂交的实验系统,找出具有抑制dot1l结合的多肽前体;
s2、通过所述多肽前体与af10的复合物进行结晶学实验,获得af10-多肽前体复合物晶体;
s3、改造所述多肽前体序列,设计出具有与af10特异性结合的多肽片段;
s4、在s3的基础上进一步优化出具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽。
从上述描述可知,本发明的有益效果在于:本发明通过结晶学实验研究dot1l酶活调节因子的结构特征进而筛选出能够特异性抑制组蛋白甲基化酶dot1l二甲基化活性的多肽前体,进而设计出能与af10特异性结合的多肽片段,科学合理且设计得到的多肽片段特异性强。
进一步地,所述步骤s1中获取af10-dot1l复合物晶体结构的操作为:鉴定af10与dot1l相互作用的区段后,通过共纯化或共表达的方式获得af10-dot1l相互作用区段的蛋白质复合物。
进一步地,所述鉴定af10与dot1l相互作用的区段的具体操作为通过二级结构预测法,利用分子克隆或大肠杆菌原核表达系统等分子生物学技术手段,共表达或分开表达出所述af10-dot1l复合物感兴趣的相互作用区段。
从上述描述可知,本发明的有益效果在于:通过精细的分子生物学实验鉴定出af10与dot1l相互作用的区段后,通过共纯化的方式获得af10-dot1l的稳定复合物。
进一步地,所述步骤s1中,所述找出具有抑制dot1l结合的多肽前体的具体操作为:建立体内蛋白互补实验、噬菌体展示以及酵母双杂交的实验系统,鉴定与af10特异结合的多肽分子。
进一步地,所述步骤s2的具体操作为:通过结晶学实验方法,对af10-抑制性多肽分子复合物的晶体生长条件进行筛选和优化,并获得能够解析该晶体结构的衍射数据。
进一步地,所述步骤s3的具体操作为:利用af10-dot1l复合物晶体结构中参与相互作用的关键氨基酸信息,改造蛋白序列,从而设计出与af10特异性结合的多肽片段。
进一步地,所述步骤s4的具体操作为:在所述步骤s3的基础上进一步通过随机突变的方式对参与af10-dot1l相互作用的关键氨基酸进行优化,优化出能与dot1l蛋白分子竞争结合af10的抑制性多肽片段。
优选地,所述具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽的氨基酸序列如seqno.1所示。
本发明还包括上述多肽在制备组蛋白去甲基化酶dot1l抑制剂药物中的应用。
从上述描述可知,本发明的有益效果在于:由于组蛋白甲基化酶dot1l二甲基化活性对白血病的发生发展起着至关重要的作用,而本发明抑制剂对于该酶具有良好的抑制作用,因此,本发明方案的抑制剂在抗癌药物中具有良好的应用前景,尤其是血癌(白血病)的治疗药物。
进一步地,所述组蛋白去甲基化酶dot1l抑制剂药物为抗癌药物。
进一步地,所述抗癌药物为抗血癌药物。
本发明的实施例一为:一种具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽的设计方法,包括以下步骤:
一、通过基于二级结构预测的方法,利用分子克隆以及大肠杆菌原核表达系统等分子生物学技术手段,共表达或者分开表达该复合物中感兴趣的相互作用区段,通过共表达或者共纯化的方法得到af10-dot1l相互作用区段的蛋白质复合物,该复合物的晶体结构如图1所示,建立体内蛋白互补实验、噬菌体展示以及酵母双杂交的实验系统,鉴定与af10特异结合的多肽分子前体。
二、通过结晶学实验方法,对af10-dot1l复合物进行晶体生长条件进行筛选和优化,获得能够解析该晶体结构的衍射数据。
三、利用af10-dot1l复合物晶体结构中参与相互作用的关键氨基酸信息,改造dot1l上的蛋白序列,从而设计出与af10特异结合的多肽片段。
四、并在此基础上进一步通过随机突变的方式对参与af10-dot1l相互作用的关键氨基酸进行优化,优化出能与dot1l蛋白分子竞争结合af10的抑制性多肽分子。
五、设计得出氨基酸序列如下的抑制性多肽:
qiewakarveklrkrnqalksqtselqrqiaeleasnaelkk,该多肽与af10的晶体结构如图2所示;通过等温滴定量热法(isothermaltitrationcalorimetry,itc)检测多肽与af10的相互作用,在恒定的温度下,配体溶液以设定的体积和次数逐滴进入样品池中。两者发生相互作用时释放或吸收的热量和两者结合的数量之间存在间接比例关系。当样品池中的大分子逐渐被滴入的配体所饱和时,相互作用产生的热量信号会逐渐减小,直到最后仅能检测到配体滴入时产生的背景稀释热量。实验条件:仪器:morocalitc200(gehealthcare),温度:25℃,样品池:af10,注射器:多肽,单次注射体积:首次注射0.4μl,随后2μl,注射时间:6s,注射间隔:180s。结果如图3所示,从图3中可以看出,本发明多肽在体外能特异性结合甲基化酶活性调节因子af10,结合常数ka达到14nm(kd=(40±10)nm;n=0.79±0.02;δs=-4.41cal/mol/deg;δh=(-11.2±0.5)kcal/mol)。结构生物学研究结果表明,如图4所示,该多肽前体对af10的结合阻断了af10与组蛋白甲基化酶dot1l二甲基化活性的能力,从而阻断了af10分子参与调节组蛋白甲基化酶dot1l二甲基化活性的能力,最终实现对该酶活性的抑制作用。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
序列表
<110>中山大学附属第五医院
<120>一种具有抑制组蛋白甲基化酶dot1l二甲基化活性的多肽、抑制剂及其设计方法与应用
<160>1
<170>siposequencelisting1.0
<210>1
<211>42
<212>prt
<213>人工序列(artificialsequence)
<400>1
glnileglutrpalalysalaargvalglulysleuarglysargasn
151015
glnalaleulysserglnthrsergluleuglnargglnilealaglu
202530
leuglualaserasnalagluleulyslys
3540
- 还没有人留言评论。精彩留言会获得点赞!
- 基于纳米颗粒团聚的dna去甲基化酶的生物传感方法
- 一种多肽——葡萄糖基转移酶蛋白-25.52和编码这种多肽的多核苷酸的制作方法
- 用于抑制法呢基蛋白转移酶的苯并(5
- 作为组蛋白脱甲基酶抑制剂的取代的(e)-n’-(1-苯基亚乙基)苯甲酰肼类似物的制作方法
- 利用雷帕霉素特异性甲基化酶标记雷帕霉素的制作方法
- 转录因子creb在调控jhdm2a基因表达及减少猪脂肪沉积中的应用的制作方法
- 一种新的多肽——dna甲基化酶13和编码这种多肽的多核苷酸的制作方法
- (3r)-去-o-甲基毛狄泼老素在制备预防或治疗抑郁症的药物中的用途的制作方法
- 法呢基蛋白转移酶的新的苯基取代的三环抑制剂的制作方法
- 新的法呢基-蛋白转移酶的三环磺酰胺抑制剂的制作方法