一种苯酞类衍生物及其制备方法和应用与流程
本发明属于有机合成化学领域,涉及一种苯酞类衍生物及其制备方法和应用。
背景技术
自然界中伞形科植物中当归属和藁本属的植物均含有苯酞类化合物,如当归和川芎等药用植物,该类化合物为植物挥发油的主要成分,大都具有浓烈的芳香气味,在植物中具有丰富的含量,且化合物结构类型较多,该类化合物普遍具有化学不稳定性,单独存在遇环境中的光、热以及酶等容易氧化或降解。但该类化合物普遍具有较好的药理活性,如抗氧化、抗血小板聚集、促进微循环、镇痛消炎、中枢神经保护以及抗肿瘤等显著的药理活性。藁本内酯是(ligustilide)传统中药当归、川芎等伞形科植物挥发油中的主要药理活性成分,其含量高达1%左右,具有显著的心脑血管药理活性,国内外学者对藁本内酯的药理活性曾经和正在进行大量的研究工作,发表了相关的药理活性研究成果,如藁本内酯对脑神经保护,改善微循环,舒张血管,抑制血管平滑肌细胞增殖,抗抑郁,抗阿尔茨海默症,镇痛消炎等重要药理作用,同时研究人员对于藁本内酯的药理研究成果进行了大量的综述(a.douglaskinghorn,heinzfalk,simongibbons,andjun’ichikobayashiedited,progressinthechemistryoforganicnaturalproducts,2017,p127,springerinternationalpublished.;左爱华,王莉,肖红斌,藁本内酯药理学和药代动力学研究进展,中国中药杂志,2012,37(22):3350-3353.)。
藁本内酯能明显减少脑缺血再灌注引起的脑梗塞体积,改善脑神经功能,并且能减轻大鼠皮层神经元和海马神经元细胞的损伤。为进一步阐明其神经保护内在作用机制,xkuang,hypeng等分别对短暂性和持久性脑缺血再灌注模型进行研究,结果表明藁本内酯呈剂量依赖性减少脑缺血脑组织中丙二醛含量,增加谷胱甘肽过氧化酶及超氧化物歧化酶活性,增强bcl-2表达同时减弱bax,caspase-3酶免疫活性,揭示出藁本内酯是通过抗氧化及抗凋亡机制对短暂性和持久性脑缺血再灌注引起的损伤起神经保护作用。在此基础上,对慢性脑缺血再灌注导致的认知缺损、生化变化及病理组织学特征进行研究,也说明藁本内酯对认知缺陷和脑损伤的保护作用是通过抗氧化及增加类胆碱功能活性实现的(xkuang,yyao,j.r.du,y.x.liu,cywang,zmqian,neuroprotectiveroleofz-ligustilideagainstforebrainischemicinjuryinicrmice,brainresearch,2006,1102(1):145-153.;haiyanpeng,junrongdu,guangyizhang,xikuang,yanxinliu,zhongmingqian,andchenyuanwang,neuroprotectiveeffectofz-ligustilideagainstpermanentfocalischemicdamageinrats,biolpharmbull,2007,30(2):309-312.)。藁本内酯通过提高细胞抗氧化能力和抑制线粒体凋亡等通路对h2o2损伤的pc12细胞起神经保护作用(yanyu,junrongdu,chenyuanwang,zhongmingqian,protectionagainsthydrogenperoxide-inducedinjurybyz-ligustilideinpc12cells,expbrainres,2008,184(3):307-312.)。hyqi等进一步研究了藁本内酯在缺氧缺糖pc12细胞中所起的双相毒物兴奋调节作用,它不仅能通过ros的生成和谷胱甘肽的消耗引起氧化应激反应,而且能通过pi3k和nrf2通路的交互作用激活促生存信号,使得藁本内酯对低、高浓度缺氧缺糖环境下的pc12细胞均能起到预先保护的作用(hongyiqi,yifanhan,jianhuirong,potentialrolesofpi3k/aktandnrf2-keap1pathwaysinregulatinghormesisofz-ligustilideinpc12cellsagainstoxygenandglucosedeprivation,neuropharmacology,2012,62(4):1659-1670.)。藁本内酯在显著改善缺血再灌注大鼠神经功能,减少梗塞体积的同时,使得炎症因子nf-κb的蛋白表达量明显减少。而且对lps致神经胶质细胞产生的tnf-α,no,il-1α,mcp-1等炎症介质有抑制作用,对α-淀粉样多肽引起的认知缺陷,神经病理学等症状,能通过调解nf-κb通路方式使其得到改善,这些都说明藁本内酯能通过抑制nf-κb通路方式起到抗炎作用。另外,藁本内酯还能通过抑制ros产生和下调mapk,nf-κb,ap-1信号通路的方式对lps致raw264.7的炎症反应起到抑制作用。总之藁本内酯也能通过抗炎机制起到神经保护的作用,这有利于神经炎症性疾病的治疗(yuwensu,wenfeichiou,shiouhueichao,menghwanlee,chienchihchen,yingchiehtsai,ligustilidepreventslps-inducedinosexpressioninraw264.7macrophagesbypreventingrosproductionanddown-regulatingthemapk,nf-κbandap-1signalingpathways,internationalimmunopharmacology,2011,11(9):1166-1172.)。
藁本内酯在1-30µm浓度时,通过p38和pi3k/akt信号通路,抑制β淀粉样蛋白诱导的神经毒作用,支持藁本内酯具有阿尔茨海默症(ad)治疗作用(weixu,liyang,jili,protectionagainstβ-amyloid-inducedneurotoxicitybynaturallyoccurringz-ligustilidethroughtheconcurrentregulationofp38andpi3-k/aktpathways,neurochemistryinternational,2016,100:44-51.)。国内外相关文献显示藁本内酯具有显著的药理活性。
苯酞类化合物根据六元环的氢化还原程度分为苯环型如正丁烯基苯酞,二氢苯环型如藁本内酯,四氢苯环型苯酞类化合物如洋川芎内酯h,同时侧链及苯环具有不同的取代情况和氧化程度,产生一系列结构多样的苯酞类化合物。同时也可以形成各种二聚和三聚体。
藁本内酯由于具有3个烯键和1个羰基双键的高度不饱和共轭体系,但未形成芳香环的稳定共轭体系,反应位点较多,反应活性较强,相对自由能较低,故容易发生1,2、1,4和1,6的diels-alder反应,形成复杂的二聚或三聚化合物。
frankr.stermita曾用芳香胺通过对内酯环的亲核取代反应,试图得到酰胺类化合物,但这种酰胺通过分子内的环加成反应过渡态最终得到环合的反应产物(johnj.beck,frankr.stermitz,additionofmethylthioglycolateandbenzylamineto(z)-ligustilide,abioactiveunsaturatedlactoneconstituentofseveralherbalmedicines.animprovedsynthesisof(z)-ligustilide,journalofnaturalproducts,1995,58(7):1047-1055.),可见藁本内酯的每个双键或官能团均具有较高的反应活性,对于以藁本内酯为原料的化学反应必须进行严格的化学反应条件控制,才能达到理想的化学反应设计。
季晖等人曾通过对于丁苯酞的化学结构改造,试图通过引入硝酸甘油的药效团提高该类化合物对血小板聚集的抑制活性和通过no抑制的抗炎活性,结果合成化合物活性比丁苯酞有了显著的提高(吴晶,凌菁菁,王旭亮,王晓丽,刘婧超,季晖,张奕华,含单硝酸异山梨醇酯的丁苯酞衍生物的合成及抗血小板聚集活性研究,中国药物化学杂志,2012,22(6):483-489.)。
国内外文献显示对于苯酞类化合物进行结构改造可以显著提高该类化合物的药理活性,但对于高度不饱和的藁本内酯,其反应条件比较苛刻,反应产物难于控制,必须对反应条件及反应催化体系进行精心的设计和把控才能达到高效合成和目标合成。药理活性研究显示,显然对于藁本内酯通过diels-alder反应建立稳定体系的化合物,药理活性相对较差,故对于藁本内酯的结构衍生重点应该放在内酯环的取代和加成优化处理方面。
丁苯酞(butylphthalide)是我国第一个拥有自主知识产权的创新药。最初是从水芹籽中提取分离得到,然后进行化学合成得到,丁苯酞具有独特的双重作用机制,既能重构微循环,增加缺血去灌注,从而保护血管结构完整、恢复血管管径、增加缺血区血流量及周围微血管数量,又能保护线粒体,减少细胞死亡,从而保护线粒体结构的完整、提高线粒体复合酶ⅳ的活性、提高线粒体atp酶的活性、维持线粒体膜的稳定性,双重狙击,对抗脑卒中。研究显示相较于藁本内酯的神经保护作用和化合物具有的毒副作用,就药理学特征丁苯酞比藁本内酯要差,原因在于显著的化学结构特征。
显而易见,由于苯酞类化合物普遍具有的化学不稳定性,虽然具有较好的药理活性,但大大限制了该类化合物在药物,保健品,食品和化妆品等领域相关产品的开发和广泛应用。因此要解决产品的开发和应用,必须首先解决该类化合物的稳定性。由于显而易见的化学结构特征,藁本内酯的药理活性要强于丁苯酞,但从药学的有效、安全和质量可控方面综合比较,丁苯酞具备药学特征(化学结构稳定),故被sfda批准上市,但目前临床反应由于其活性和毒副作用等原因,临床医生已经对丁苯酞提出进一步优化和提高活性的强烈需求,并不能满足临床对于脑卒中的治疗作用要求。因此有必要针对藁本内酯的独特化学结构进行结构的人为干预,优化和筛选高效和稳定的基于该类化合物的心脑血管治疗药物。
技术实现要素:
本发明的目的在于提供一种苯酞类衍生物及其制备方法和应用。该苯酞类衍生物的显著特征在于其原有的内酯结构片段被内酰胺基团所取代,c-3位被羟基取代。
一种苯酞类衍生物,其特征在于该衍生物的结构如下:
如上所述苯酞类衍生物的制备方法,具体步骤为:将苯酞类化合物加入有机溶剂溶解,温度控制在-20℃-60℃,加入有机溶剂溶解的环丙胺反应液,控制温度在-20℃-60℃,搅拌反应1-24h,减压回收有机溶剂,经重结晶得到目标产物。
所述苯酞类化合物与环丙胺的摩尔比为1:1~1.2。
所述苯酞类化合物为藁本内酯、正丁烯苯酞以及含藁本内酯或正丁烯苯酞的药材挥发油提取物中的一种或两种以上。
所述药材挥发油提取物为当归挥发油提取物或川芎挥发油提取物。
所述有机溶剂为非极性有机溶剂,优选环己烷、石油醚、四氢呋喃等。
所述重结晶所用溶剂为石油醚、乙酸乙酯、丙酮中的一种或两种。
如上所述苯酞类衍生物在抗心脑血管疾病和抗氧化方面的应用。
所述苯酞类衍生物为式ⅰ化合物和式ⅱ化合物的组合物,组合物中式ⅰ化合物的质量百分含量为1-99%。
所述式ⅰ化合物的质量百分含量为90%以上。
本发明中采用化学结构衍生方法合成的苯酞类衍生物的化学结构均通过质谱(hr-esi-ms),核磁(1d,2d-nmr),单晶衍射(x-ray),液相色谱(hplc-dad)等多种检测技术证明为目标化合物。
本发明中苯酞类衍生物通过体内外药理活性研究证明具有较好的抗氧化神经细胞保护和抗血小板聚集作用,可以作为心脑血管疾病的化学预防和治疗药物的应用。
本发明的优点在于:
1、本发明的主体化合物属于异吲哚酮类化合物,式ⅰ化合物在非手性合成反应条件下,通过分子内的自身环化亲核取代反应,生成c-3位具有消旋的立体对应异构体,再通过重结晶简单拆分方法直接得到c-3位羟基为s构型的单一对映体的手性化合物。
2、借助环丙胺的独特化学结构和显著的药效集团,通过化学结构衍生构建和制备的苯酞类衍生物化学结构稳定,有效增强了天然或有机全合成苯酞类化合物的稳定性,增加了该类化合物的成药性,有效掩盖该类化合物的不良挥发性气味。
3、通过化学结构衍生制备的苯酞类衍生物形成独特的立体化学结构,增强了该类化合物与药物作用靶点的空间结合度,有效增强了该类化合物的药理活性。该类化合物反应中,首先环丙胺与藁本内酯环中的羰基进行亲核取代反应,形成酰胺和1-戊酮的开环关键中间体,该中间体中环丙烷中的氮原子具有较强的亲核反应性能,继续与1-戊酮基的酮羰基进行亲核加成反应,但此时环丙胺中的环丙烷基团可以看作是一个高度共轭的平面结构,由于空间位阻的影响,羰基碳与环丙胺中的氮形成的碳氮键进行自由旋转受到抑制,力求减小立体空间位阻,促使1-戊酮基上的亲核加成反应发生,结果环丙烷和羟基的立体结构和空间体积比较小,同时偏向于内酰胺和己二烯形成的平面结构的同一侧,而空间体积较大的4碳脂肪烷侧链趋向于另一侧,因此苯酞类化合物与环丙胺反应形成独特的立体化学结构反应产物。
4、通过本发明的结构改造得到苯酞类化合物,丰富了该类化合物的化学结构库,为该类化合物的药物筛选提供大量的先导化合物。
附图说明
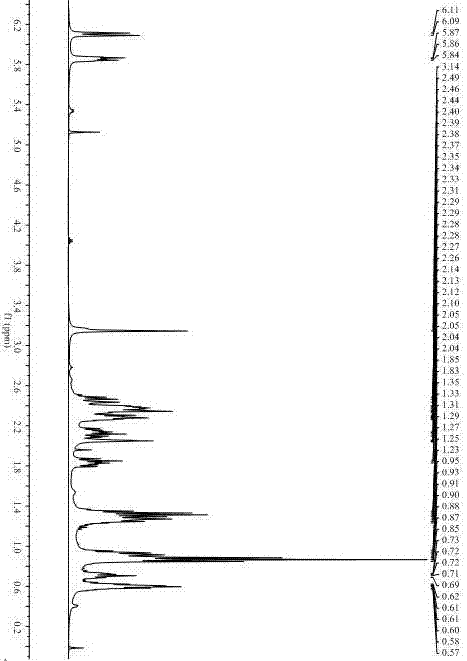
图1化合物n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的1h-nmr图谱。
图2化合物n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的13c-nmr图谱。
图3化合物n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的dept图谱。
图4化合物n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的1h-1hcosy图谱。
图5化合物n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的hsqc图谱。
图6化合物n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的hmbc图谱。
图7化合物n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的单晶衍射(x-ray)结构,图a为单晶结构,图b为晶胞内双分子单晶结构,图示c-3位立体构型为s构型的单一对映体。
图8化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的单晶x-ray衍生结构图;图a为单晶结构,图b为晶胞内双分子单晶单晶结构,图示c-3位立体构型为r和s构型同时存在的外消旋体。
图9化合物n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的hplc-dad色谱检测结果,图中tr=14.863min处色谱峰为目标化合物。检测仪器:安捷伦1200色谱系统配备dad检测器;色谱条件:色谱柱,xterramsc18(waters);填料粒径5µm,柱长4.6×250mm。流动相:甲醇:水=65:35(v/v)。手动进样器进样,每次20µl。检测波长280nm。
图10化合物n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺和化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的二者混合样品hplc-dad色谱检测结果,图中tr=13.840min处色谱峰为n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的色谱峰。tr=15.348min处色谱峰为n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的色谱峰。色谱条件同图9色谱条件。
图11dad检测化合物n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的uv光谱图。
图12dad检测化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的uv光谱图。
图13化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的hplc-dad手性拆分色谱图。检测仪器:安捷伦1200色谱系统配备dad检测器;色谱条件:色谱柱,chiralcelod-h手性涂敷柱;填料粒径5µm,柱长4.6×150mm。流动相:正己烷:异丙醇=98:2(v/v)。手动进样器进样,每次20µl。检测波长283nm。
图14苯酞及其衍生物对于过氧化氢诱导pc12神经细胞氧化损伤的保护作用。其中“**”表示统计学上p<0.01%,具有较显著差异;“*”表示统计学上p<0.05%,具有显著差异。
具体实施方式
实施例1:n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺的合成。
将藁本内酯(1.900g,0.010mol)溶解在50ml的四氢呋喃中,环丙胺(0.684g,0.012mol)溶解在20ml的四氢呋喃中,水浴控制温度在25℃,机械搅拌下,将环丙胺的四氢呋喃溶液点滴加入到藁本内酯的四氢呋喃溶液中,搅拌反应4h,旋蒸减压回收四氢呋喃和过量的环丙胺溶液,将200ml石油醚加入到以上浓缩物中,混合均匀,放置过夜待合成产物结晶洗出,抽滤,得粗产物1.976克,反应产率80%,将粗产物用石油醚和丙酮(5:1,v/v)的混合溶剂重结晶,得到结晶产物0.97g,结晶产率49%。采用hplc-dad方法,面积归一化含量测定目标化合物的含量达到99.5%(见附图9)。目标化合物通过ms,nmr和x-ray单晶衍射技术检测为目标化合物(见附图1-7)。
n-环丙烷基-3-正丁基-3-s-羟基-藁本内酰胺:无色针状或棱柱状结晶,hr-esi-msm/z270.1465[m+h]+(计算值c15h21nnao2为270.1470),1h-nmr(400mhz,acetone-d6):δ6.11(1h,dt,j=12.0,4.0hz,h-7),5.86(1h,dt,j=8.0,4.0,hz,h-6),2.49-2.43(1h,m,h-4),2.41-2.34(1h,m,h-4),2.39-2.36(2h,m,h-5),2.31-2.27(1h,m,h-12),2.17-2.11(1h,m,h-8),1.88-1.82(1h,m,h-8),1.35-1.31(2h,m.h-10),1.28-1.22(1h,m,1.27-1.25(1h,m,h-13),0.96-0.90(2h,m.h-9),0.88-0.85(3h,t,j=8.0hz,h-11),0.75-0.69(1h,m,h-14),0.64-0.58(1h,m,h-13),0.63-0.58(1h,m,h-14);13c-nmr(100mhz,acetone-d6):δ169.2(c-1),152.6(c-3a),128.9(c-7a),128.3(c-6),117.4(c-7),92.4(c-3),33.9(c-8),25.9(c-9),22.6(c-10),21.1(c-12),13.7(c-11),5.1(c-14),2.7(c-13)。
晶胞参数:正交晶系,空间群p212121,a=7.8762(15),b=10.576(2),c=16.809(3),α=β=γ=90°,_晶胞体积v=1400.1(5),晶胞内分子数z=4,采用仪器:brukerapexii面检测仪,照射波长采用0.71073,放射源为moka,收集2876个独立衍射点,其中有效衍射点2037个,参加精修衍射点数目:2876;参加参数数目:165,对于全部衍射点的r1值:0.0775;对于可观测衍射点的r1值:0.0531;对于全部衍射点的wr2值:0.1348;对于可观测衍射点的wr2值:0.1203;对于可观测衍射点的s值:1.003;对于全部衍射点的s值:1.003;最后精修过程中的最大移动值:0.000。
实施例2:n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的合成。
将藁本内酯(1.880g,0.010mol)溶解在50ml的四氢呋喃中,环丙胺(0.684g,0.012mol)溶解在20ml的四氢呋喃中,常温机械搅拌下,将环丙胺的四氢呋喃溶液点滴加入到藁本内酯的四氢呋喃溶液中,搅拌反应6h,旋蒸减压回收四氢呋喃和过量的环丙胺溶液,将200ml石油醚加入到以上浓缩物中,混合均匀,放置过夜待合成产物结晶洗出,抽滤,得粗产物2.083克,反应产率85%,将粗产物用石油醚和丙酮(10:1,v/v)的混合溶剂重结晶,得到结晶产物1.666g,结晶产率80%。采用hplc-dad方法,面积归一化含量测定目标化合物的含量达到99.6%。目标化合物通过ms,nmr和x-ray单晶衍射技术检测为目标化合物(见附图8,10,11,12),通过高效液相色谱的手性拆分确定该化合物为一对手性对应异构体(见图13)。
n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺:无色针状或棱柱状结晶,hr-esi-msm/z246.1492[m+h]+(计算值c15h20no2为246.1489),1h-nmr(400mhz,acetone-d6):δ7.63(1h,d,j=8.0hz,h-7),7.60(1h,t,j=8.0,4.0hz,h-5),7.55(1h,d,j=8.0hz,h-4),7.49(1h,t,j=8.0,4.0hz,h-4),2.45-2.50(1h,td,j=8.0,4.0hz,h-8),2.38-2.38(1h,td,j=12.0,4.0hz,h-8),2.25-2.16(1h,td,j=16.0,4.0hz,h-9),2.05-2.07(1h,m,h-12),1.49-1.42(1h,m,h-9),1.30-1.18(2h,m,h-10),0.89-1.00(1h,m,h-13),0.82-0.87(1h,m,h-13),0.79(3h,t,j=16.0,8.0hzh-11),0.58-0.76(2h,m,h-14);13c-nmr(100mhz,acetone-d6):δ166.2,145.8,130.4,130.0,127.3,120.6,120.2,90.2,34.4,24.0,20.5,19.7,11.6,3.20,0.90。
实施例3:采用当归挥发油进行化合物n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺和化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺二者混合物的合成
将制备得到的当归挥发油采用柱色谱法,以石油醚和乙酸乙酯(3:1,v/v)为洗脱剂进行预分离,将含藁本内酯较高部分的当归挥发油(3.000g,)溶解在60ml的四氢呋喃中,环丙胺(0.684g,0.012mol)溶解在30ml的四氢呋喃中,机械搅拌下,将环丙胺的四氢呋喃溶液点滴加入到以上当归挥发油的四氢呋喃溶液中,搅拌反应10h,旋蒸减压回收四氢呋喃和过量的环丙胺溶液,将200ml石油醚加入到以上浓缩物中,混合均匀,放置过夜待合成产物结晶洗出,抽滤,得粗产物2.283克,反应产率76%,将粗产物用石油醚和丙酮(10:1,v/v)的混合溶剂重结晶,得到结晶产物1.872g,结晶产率82%。采用hplc-dad方法,面积归一化含量测定目标化合物的含量:化合物n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺的含量90.0%;化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的含量为8.9%。
实施例4:采用川芎挥发油进行化合物n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺和化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺二者混合物的合成
将制备得到的川芎挥发油采用柱色谱法,以石油醚和乙酸乙酯(3:1,v/v)为洗脱剂进行预分离,将含藁本内酯较高部分的川芎挥发油(3.500g,)溶解在60ml的四氢呋喃中,环丙胺(0.684g,0.012mol)溶解在30ml的四氢呋喃中,机械搅拌下,将环丙胺的四氢呋喃溶液点滴加入到以上川芎挥发油的四氢呋喃溶液中,搅拌反应10h,旋蒸减压回收四氢呋喃和过量的环丙胺溶液,将200ml石油醚加入到以上浓缩物中,混合均匀,放置过夜待合成产物结晶洗出,抽滤,得粗产物1.982克,反应产率56.7%,将粗产物用石油醚和丙酮(10:1,v/v)的混合溶剂重结晶,得到结晶产物1.685g,结晶产率85%。采用hplc-dad方法,面积归一化含量测定目标化合物的含量:化合物n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺的含量为91.2%;化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的含量为7.9%。
实施例5:采用当归挥发油进行n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺和化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺单体化合物的合成制备
将制备得到的当归挥发油采用柱色谱法,以石油醚和乙酸乙酯(3:1,v/v)为洗脱剂进行预分离,将含藁本内酯较高部分的当归挥发油(3.000g,)溶解在60ml的四氢呋喃中,环丙胺(0.684g,0.012mol)溶解在30ml的四氢呋喃中,机械搅拌下,将环丙胺的四氢呋喃溶液点滴加入到以上当归挥发油的四氢呋喃溶液中,水浴加热至45℃,恒温搅拌反应10h,旋蒸减压回收四氢呋喃和过量的环丙胺溶液,将200ml石油醚加入到以上浓缩物中,混合均匀,放置过夜待合成产物结晶洗出,抽滤,得粗产物2.562克,反应产率85.4%,将粗产物用石油醚和丙酮(10:1,v/v)的混合溶剂重结晶,得到结晶产物2.050g,结晶产率80%。采用hplc-dad方法,面积归一化含量测定目标化合物的含量:化合物n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺的含量为70.0%;化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的含量为28.1%(见附图5、图6、图7)。
实施例6:采用当归挥发油进行n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺和化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺单体化合物的合成制备
将制备得到的当归挥发油采用柱色谱法,以石油醚和乙酸乙酯(3:1,v/v)为洗脱剂进行预分离,将含藁本内酯较高部分的当归挥发油(3.000g,)溶解在60ml的四氢呋喃中,环丙胺(0.684g,0.012mol)溶解在30ml的四氢呋喃中,机械搅拌下,将环丙胺的四氢呋喃溶液点滴加入到以上当归挥发油的四氢呋喃溶液中,常温机械搅拌反应10h,旋蒸减压回收四氢呋喃和过量的环丙胺溶液,将200ml石油醚加入到以上浓缩物中,混合均匀,放置过夜待合成产物结晶洗出,抽滤,得粗产物2.302克,反应产率76.7%。
将粗产物用石油醚和乙酸乙酯(3:1,v/v)为洗脱液,进行柱色谱分离纯化,分别得到a、b和c三个组分,得到组份a(0.483g),经薄层色谱鉴定为n-环丙烷基-3-正丁基-3-苯酞内酰胺;得到组份b(0.420g)经薄层色谱鉴定为化合物n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺和化合物n--环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的混合物;得到组份c(1.128g)。经薄层色谱鉴定为化合物n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺;采用hplc-dad方法,面积归一化进行目标化合物的含量测定:组份a为99.4%;组份b为:化合物n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺的含量为66.9%,化合物n--环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的含量为36.5%;组份c为99.6%。
实施例7:采用当归和川芎的挥发油混合物进行化合物n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺和化合物n-环丙烷基-3-正丁基-3-羟基-苯酞内酰胺化合物的合成
将制备得到的当归和川芎的挥发油混合物采用柱色谱法,以石油醚和乙酸乙酯(3:1,v/v)为洗脱剂进行预分离,将含藁本内酯较高部分的挥发油混合物(3.000g,)溶解在60ml的四氢呋喃中,环丙胺(0.684g,0.012mol)溶解在30ml的四氢呋喃中,机械搅拌下,将环丙胺的四氢呋喃溶液点滴加入到以上当归挥发油的四氢呋喃溶液中,常温搅拌反应6h,旋蒸减压回收四氢呋喃和过量的环丙胺溶液,将200ml石油醚加入到以上浓缩物中,混合均匀,-20℃放置过夜待合成产物结晶洗出,抽滤,得粗产物2.602克,反应产率86.7%,将粗产物用石油醚和丙酮(10:1,v/v)的混合溶剂重结晶,得到结晶产物2.080g,结晶产率80%。采用hplc-dad方法,面积归一化含量测定目标化合物的含量:化合物n-环丙烷基-3-正丁基-3-羟基-藁本内酰胺的含量为90.5%;化合物n--环丙烷基-3-正丁基-3-羟基-苯酞内酰胺的含量为27.8%。
实施例8:苯酞类化合物神经保护作用研究。
1、材料:pc12细胞(中科院上海细胞所);苯酞及其衍生物(本专利申请人实验室制备,hplc-dad面积归一化法测定其含量达到98%),高糖培养基(dmem,南京凯基生物科技发展有限公司);二甲基亚砜(dmso,sigma公司);胎牛血清(gibco,赛默飞世尔科技有限公司);h2o2(南京化学试剂股份有限公司);噻唑蓝(mtt,南京生兴生物技术有限公司),酶标仪(美国bio-rad公司)。
2、方法:pc12细胞培养,取pc12细胞接种于高糖dmem培养基中,培养基中含有10%胎牛血清、青霉素100uml-1,链霉素100µgml-1,置于37℃、5%的co2细胞培养箱中培养,隔日换液,常规胰蛋白酶消化传代;待细胞长至70-80%时用于实验。
3、苯酞及其衍生物对pc12细胞增殖的影响:取对数生长期的pc12细胞以1×104细胞/孔密度接种于96孔细胞培养板中,孵育24h后,加入100µl苯酞及其衍生物溶液,使其终浓度分别为1.25、2.5、5.0、10、20、40、80、160µm,继续培养24h,加入100µl的mtt(5mgml-1),孵育4h后,加入150µl的dmso,振荡10min后,选择490nm波长,检测各孔吸光度(a)值。
细胞存活率(%)=(a实验组–a空白组)/(a对照组-a空白组)×100%。
结果显示200µm以上的藁本内酯,藁本内酯衍生物,正丁烯苯酞、正丁烯苯酞衍生物才会对pc12细胞的生长受到抑制。
4、氧化损伤模型建立:取对数生长期的pc12细胞以5×103细胞/孔密度接种于96孔细胞培养板中,加入不同浓度的h2o2,培养4h,按以上mtt测定法项下操作加入mtt,测定各孔a值,最终选择降低细胞存活率50%的h2o2浓度为造模浓度。
结果显示250µm的h2o2可以使50%的pc12细胞生长受到抑制。故选择250µm作为神经细胞氧化受损的诱导浓度。
5、分组及药物处理:实验分组为正常空白对照组、h2o2模型组(250µm)、苯酞类及其衍生物。(2.5µm,5.0µm,10µm,25µm,50µm)浓度组、阳性对照组(丁苯酞)。苯酞类及其衍生物各组和阳性对照组分别加入h2o2250μm。实验首先分别加入苯酞类及其衍生物溶液,使其终浓度达到相应值,4h后加入h2o2,24h后加入mtt,检测a值,计算细胞存活率。
研究结果见附图8,图中“**”表示该结果与阳性对照比较有统计学较显著性差异p<0.01%,图中“*”表示该结果与阳性对照比较有统计学显著性差异p<0.05%。图中可以看出,藁本内酯衍生物的对于pc12神经细胞的过氧化氢保护作用最强,在最低实验难度2.5µm时,可以使细胞活度提高80%,相较于模型组的52%和阳性药物对照组的68%,该化合物可以显著保护过氧化氢对于pc12的氧化损伤。藁本内酯衍生物的浓度在10µm时,细胞活度最高,达到95%。其次藁本内酯的活性较正丁烯苯酞对于神经细胞的氧化损伤保护作用较强;藁本内酯与正丁烯苯酞衍生物对于神经细胞的氧化损伤保护作用几乎相同。
- 还没有人留言评论。精彩留言会获得点赞!