一种含有萘并双噻二唑结构的有机光电材料及其应用的制作方法
本发明属于有机光电材料技术领域,尤其涉及一种含有萘并双噻二唑结构的有机光电材料及其应用。
背景技术:
随着信息时代的来临,新型显示器件的研制越来越引起人们的重视,特别是各类平板显示器件(FPD)以其体积小、重量轻、能耗低、屏幕大等特点,引发了一股强劲的平板显示器件研制热潮。作为新一代平板显示器件,有机电致发光器件(OLED)在手机、PDA、数码相机、车载显示、笔记本电脑、壁挂电视以及军事领域都具有广阔的应用前景,是一种将来替代液晶显示器(LCD)的新型平板显示器件。正因如此,OLED是近几年来新材料及显示技术领域研究、开发的一大热点,其产业化势头十分迅猛。目前,国内外对OLED的研究主要集中在发光材料的研究、器件的制作和产品开发上。有机电致发光器件(organic light emission devices,OLED)具有如下的特点:(1)采用有机物,材料选择范围宽,可实现从蓝光到红光的任何颜色的显示;(2)驱动电压低,只需3---10V的直流电压;(3)发光亮度和发光效率高;(4)全固化的主动发光;(5)视角宽,响应速度快(微秒量级);(6)制备过程简单,费用低;(7)超薄,重量轻;(8)可做在柔性衬底上,器件可弯曲,折叠。因此,有机电致发光器件可应用于室内照明,各种显示屏,坦克、飞机等现代化武器中的显示终端等。有机电致发光也可应用野外照明;可制成光电藕合器,用于光通讯,即用作集成电路上的芯片与芯片之间的单片光源;可制作可折叠的“电子报纸”;可应用于飞机、坦克等的数字、图象处理和移动通信装置的显示,它在彩色大屏幕平板显示技术方面已经显示出了广阔的应用前景。它能克服液晶显示的视角小、响应速度慢、等离子显示的高电压以及无机电致发光的发光品种少的缺点,成为取代传统阴极射线显像管的平板显示技术的有力竞争者。正是由于有机发光器件的诸多优点以及广阔的应用前景,目前国内外约80多家大公司在从事有机电致发光材料和器件的研究开发工作。
1988年C.Adachi等人首次提出了将空穴传输层、电子传输层和发光层分开的三层结构,获得了高亮度和长寿命的蓝光器件;1994年在日本滨松召开的有机及无机电致发光国际会议上,C.W.Tang首次报道了使用寿命达10000小时的双层结构有机电致发光器件;1998年,美国普林斯顿大学的Forrest小组首次提出将磷光染料应用于有机电致发光器件,这样就突破了器件内量子效率低于25%的限制,理论上使内量子效率达到了100%,从而开创了有机磷光电致发光的新领域。同年,T.R.Hebner等发明了制备有机电致发光器件的喷墨打印法,这为有机电致发光器件从研究走向市场提供了更大的可能。进入新世纪,各种新型有机发光器件更是层出不穷,比较有代表性的是:2004年,L.S.Liao等人制作的叠层(tandem)有机电致发光器件,电流效率高达136cd/A。
目前的研究中有机电荷传输材料的发展是制约有机电致发光器件发展的瓶颈,合成和筛选较好的有机小分子电荷传输材料对提高OLED器件的发光效率具有重要意义。
技术实现要素:
本发明为了解决上述技术问题提供一种含有萘并双噻二唑结构的有机光电材料,其可有效的应用于有机电致发光器件中,以此提高发光器件的光电性能。
本发明解决上述技术问题的技术方案如下:一种含有萘并双噻二唑结构的有机光电材料,包括具有符合式Ⅰ所示的分子结构:
其中Ar1、Ar2相同或不相同,所述Ar1、Ar2选自氢、取代或未取代C4-C36且含有N、O中至少一种的芳族杂环基中的一种。
进一步,所述Ar1、Ar2选自下述基团中的任意一种:
其中,*表示键合位点。
进一步,所述式Ⅰ的具体化合物结构式为下述化合物C01-C41中的一种:
本发明提供一种含有萘并双噻二唑结构的有机光电材料在有机电致发光器件中的应用。
本发明还提供一种有机电致发光器件,包括至少一层功能层,所述功能层含有上述含有萘并双噻二唑结构的有机光电材料。
进一步,所述一种有机电致发光器件,包括发光层、空穴传输层与电子传输层,所述发光层、空穴传输层或电子传输层含有上述含有萘并双噻二唑结构的有机光电材料。
本发明还提供一种照明或显示原件,包括上述所述的有机电致发光器件。
本发明的有益效果是:
本发明含有萘并双噻二唑结构的有机光电材料应用于有机电致发光器件中获得了高效的电致发光性能,其主要优点如下:
1、该类材料分子空间结构为扭曲非平面结构,有效避免了分子紧密积聚,具有较高的荧光量子效率。
2、具有优异的热稳定性,玻璃化转变温度和分解温度都很高,容易形成良好的无定形薄膜,应用在电致发光器件中,可以获得更加稳定的效果和更长的使用寿命。
3、该类材料具有适合的分子能级,适合作蓝色发光材料的的主体材料,同时具有红色和绿色应用的潜力,因此即可应用于红绿蓝三基色掺杂性主体材料,也可单独作为空穴传输材料、电子传输材料或者发光层。
附图说明
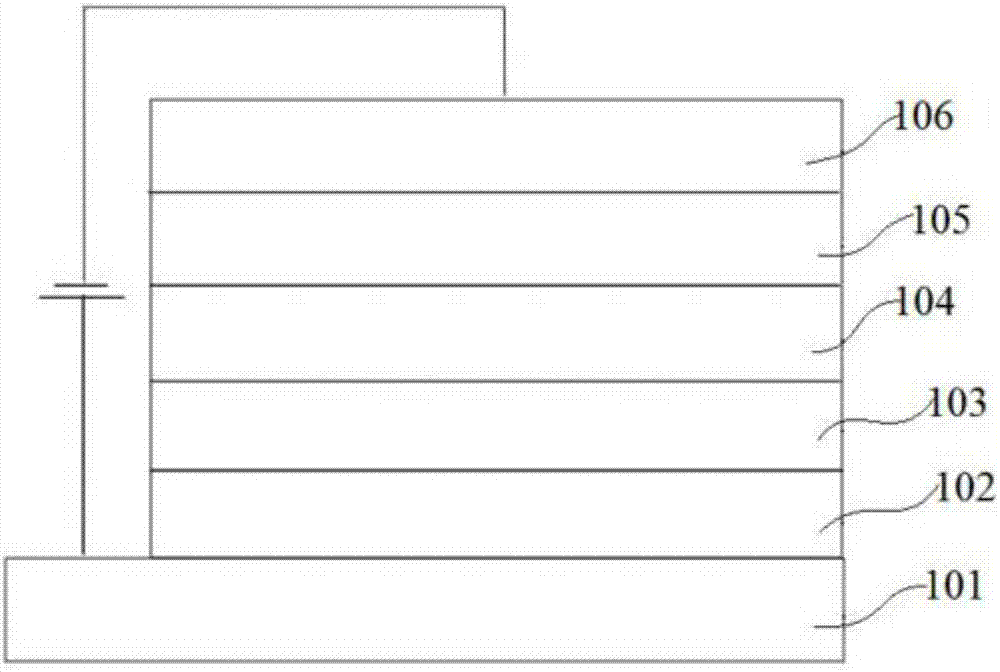
图1为本发明制备的有机电致发光器件结果示意图。
附图中,各标号所代表的含义如下:
101、阳极层,102、空穴传输层,103、发光层,104、电子传输层,105、电子注入层,106、阴极层。
具体实施方式
以下对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
在下述实施例中,所使用到的试剂、材料以及仪器如没有特殊的说明,均为常规试剂、常规材料以及常规仪器,均可商购获得,其中所涉及的试剂也可通过常规合成方法合成获得。
实施例一化合物的制备
本实施例给出了实施例一种的C01~C41中的部分化合物的制备方法。
实施例1化合物C01的制备
(1)中间体1的制备
中间体1的制备:在1L三口瓶中,加入原料萘并噻二唑(18.6g,0.1mol)和500g氯仿,室温搅拌,慢慢滴入浓硝酸和浓硫酸的混合液,约15min滴加完毕,室温搅拌反应14小时。将以上反应液分液,收集有机相,减压脱去溶剂,得到22.4g黄色固体,收率97%。
DEI-MS,分子式C10H5N3O2S,理论值231.01,测试值231.65。
(2)中间体2的制备
中间体2的制备:在1L三口瓶中,加入中间体1(11.6g,0.05mol)和350mL热乙醇,搅拌溶解后加入盐酸羟胺(2.6g,0.05mol),将反应体系快速转移至低温浴槽中冷却至-15℃,滴加KOH(28.0g,0.5mol)的乙醇(200mL)溶液,控制内温低于-10℃,滴加完毕后恢复至室温继续反应10小时。将以上反应液慢慢倾入1.5L冰水中,有绿色固体析出,抽滤,收集滤饼,获得11.3g绿色粉末,收率为92%。
DEI-MS,分子式C10H6N4O2S,理论值246.02,测试值246.51。
(3)中间体3的制备
中间体3的制备:在1L三口瓶中,加入中间体2(12.3g,0.05mol)和300g水,混合物加热至回流状态,加入保险粉(34.8g,0.2mol),体系变为深红色,继续反应2小时。混合物趁热过滤,滤液冷却至室温,有红色固体析出,抽滤,收集滤饼,获得6.7g红色粉末,收率为62%。
DEI-MS,分子式C10H8N4S,理论值216.05,测试值216.33。
(4)中间体4的制备
中间体4的制备:在500mL三口瓶中,加入中间体3(4.3g,0.02mol)、100g干燥的氯仿和3g吡啶,慢慢滴加20mL二氯亚砜和10mL氯仿的混合溶液,约15min滴加完毕,升温至回流状态搅拌反应1小时。将以上反应液减压脱去溶剂,得到棕色固体,加入200g冰水继续搅拌1小时,抽滤,收集滤饼,获得4.9g棕红色粉末,收率为100%。
DEI-MS,分子式C10H4N4S2,理论值243.99,测试值244.26。
(5)中间体5的制备
中间体5的制备:在500L三口瓶中,加入中间体4(2.4g,0.01mol)和200mL48%氢溴酸,慢慢滴入3.2g(0.02mol)溴素,约10min滴加完毕,继续搅拌反应24小时,抽滤,收集滤饼,获得2.1g红色粉末,收率为51%。
DEI-MS,分子式C10H2Br2N4S2,理论值399.81,测试值399.69。
(6)化合物C01的制备
化合物C01的制备:在1L三口瓶中,氮气保护下,加入中间体5(4.0g,0.01mol),咔唑(3.7g,0.022mol)、碳酸钾(4.2g,0.03mol)和500g邻二氯苯,氮气保护下加入催化剂CuI(0.4g,0.002mol)和邻菲啰啉(0.7g,0.004mol),控内温160~165℃,保温反应18.0hrs后,降至20~25℃,抽滤,滤液减压脱溶剂至无馏分,过硅胶柱,得到3.9g目标产物,纯度99.56%,收率为68.4%。
DEI-MS,分子式C34H18N6S2,理论值574.12,测试值574.01。
实施例2化合物C03的制备
化合物C03的制备:在1L三口瓶中,氮气保护下,加入中间体5(4.0g,0.01mol),化合物A1(7.4g,0.022mol)、碳酸钾(4.2g,0.03mol)和500g邻二氯苯,氮气保护下加入催化剂CuI(0.4g,0.002mol)和邻菲啰啉(0.7g,0.004mol),控内温160~165℃,保温反应18.0hrs后,降至20~25℃,抽滤,滤液减压脱溶剂至无馏分,过硅胶柱,得到5.8g目标产物,纯度99.36%,收率为63.5%。
DEI-MS,分子式C58H36N8S2,理论值908.25,测试值908.33。
实施例3化合物C04的制备
化合物C04的制备:在1L三口瓶中,氮气保护下,加入中间体5(4.0g,0.01mol),化合物A2(7.3g,0.022mol)、碳酸钾(4.2g,0.03mol)和500g邻二氯苯,氮气保护下加入催化剂CuI(0.4g,0.002mol)和邻菲啰啉(0.7g,0.004mol),控内温160~165℃,保温反应18.0hrs后,降至20~25℃,抽滤,滤液减压脱溶剂至无馏分,过硅胶柱,得到5.4g目标产物,纯度99.58%,收率为59.3%。
DEI-MS,分子式C58H32N8S2,理论值904.22,测试值904.86。
实施例4化合物C09的制备
化合物C09的制备:在1L三口瓶中,氮气保护下,加入中间体5(4.0g,0.01mol),化合物A3(5.7g,0.022mol)、碳酸钾(4.2g,0.03mol)和500g邻二氯苯,氮气保护下加入催化剂CuI(0.4g,0.002mol)和邻菲啰啉(0.7g,0.004mol),控内温160~165℃,保温反应18.0hrs后,降至20~25℃,抽滤,滤液减压脱溶剂至无馏分,过硅胶柱,得到4.6g目标产物,纯度99.48%,收率为61.3%。
DEI-MS,分子式C46H26N6O2S2,理论值758.16,测试值758.66。
实施例5化合物C15的制备
化合物C15的制备:在1L三口瓶中,氮气保护下,加入中间体5(4.0g,0.01mol),化合物A4(6.3g,0.022mol),随后加入100mL甲苯和50mL碳酸钠水溶液(2mol/L)。氮气保护下,回流搅拌加入Pd(PPh3)4(30mg,0.05mmol),回流反应12小时。反应完毕后向体系加入100mL水,分液。有机相脱溶剂后得到粗品,过硅胶柱,得到4.9g目标产物,纯度99.44%,收率为67.6%。
DEI-MS,分子式C46H26N6S2,理论值726.17,测试值726.36。
实施例6化合物C19的制备
化合物C19的制备:在1L三口瓶中,氮气保护下,加入中间体5(4.0g,0.01mol),化合物A5(7.2g,0.022mol),随后加入100mL甲苯和50mL碳酸钠水溶液(2mol/L)。氮气保护下,回流搅拌加入Pd(PPh3)4(30mg,0.05mmol),回流反应12小时。反应完毕后向体系加入100mL水,分液。有机相脱溶剂后得到粗品,过硅胶柱,得到5.8g目标产物,纯度99.51%,收率为72.1%。
DEI-MS,分子式C52H38N6S2,理论值810.26,测试值810.54。
实施例7化合物C20的制备
化合物C20的制备:在1L三口瓶中,氮气保护下,加入中间体5(4.0g,0.01mol),化合物A6(6.7g,0.022mol),随后加入100mL甲苯和50mL碳酸钠水溶液(2mol/L)。氮气保护下,回流搅拌加入Pd(PPh3)4(30mg,0.05mmol),回流反应12小时。反应完毕后向体系加入100mL水,分液。有机相脱溶剂后得到粗品,过硅胶柱,得到5.3g目标产物,纯度99.61%,收率为69.5%。
DEI-MS,分子式C46H26N6O2S2,理论值758.16,测试值758.54。
实施例8化合物C25的制备
(1)中间体6的制备
中间体6的制备:在1L三口瓶中,氮气保护下,加入中间体5(4.0g,0.01mol),化合物A3(2.9g,0.011mol)、碳酸钾(2.1g,0.015mol)和500g邻二氯苯,氮气保护下加入催化剂CuI(0.4g,0.002mol)和邻菲啰啉(0.7g,0.004mol),控内温160~165℃,保温反应18.0hrs后,降至20~25℃,抽滤,滤液减压脱溶剂至无馏分,过硅胶柱,得到3.6g目标产物,纯度99.48%,收率为61.3%。
DEI-MS,分子式C28H14BrN5OS2,理论值578.98,测试值578.99。
(2)化合物C25的制备
化合物C25的制备:在1L三口瓶中,氮气保护下,加入中间体6(5.8g,0.01mol),化合物A4(3.2g,0.011mol),随后加入100mL甲苯和50mL碳酸钠水溶液(2mol/L)。氮气保护下,回流搅拌加入Pd(PPh3)4(30mg,0.05mmol),回流反应12小时。反应完毕后向体系加入100mL水,分液。有机相脱溶剂后得到粗品,过硅胶柱,得到5.5g目标产物,纯度99.63%,收率为73.6%。
DEI-MS,分子式C46H26N6OS2,理论值742.16,测试值742.44。
实施例9化合物C26的制备
化合物C26的制备:在1L三口瓶中,氮气保护下,加入中间体6(5.8g,0.01mol),化合物A7(3.2g,0.011mol),随后加入100mL甲苯和50mL碳酸钠水溶液(2mol/L)。氮气保护下,回流搅拌加入Pd(PPh3)4(30mg,0.05mmol),回流反应12小时。反应完毕后向体系加入100mL水,分液。有机相脱溶剂后得到粗品,过硅胶柱,得到4.0g目标产物,纯度99.53%,收率为53.6%。
DEI-MS,分子式C46H26N6OS2,理论值742.16,测试值742.48。
实施例10化合物C35的制备
(1)中间体7的制备
中间体7的制备:在500L三口瓶中,加入中间体4(2.4g,0.01mol)和200mL48%氢溴酸,慢慢滴入1.6g(0.01mol)溴素,约10min滴加完毕,继续搅拌反应24小时,抽滤,收集滤饼,过硅胶柱,获得1.6g红色粉末,收率为51%。
DEI-MS,分子式C10H3BrN4S2,理论值321.90,测试值321.99。
(2)化合物C35的制备
化合物C35的制备:在1L三口瓶中,氮气保护下,加入中间体7(3.2g,0.01mol),化合物A8(3.0g,0.011mol)、碳酸钾(4.2g,0.03mol)和500g邻二氯苯,氮气保护下加入催化剂CuI(0.4g,0.002mol)和邻菲啰啉(0.7g,0.004mol),控内温160~165℃,保温反应18.0hrs后,降至20~25℃,抽滤,滤液减压脱溶剂至无馏分,过硅胶柱,得到3.8g目标产物,纯度99.66%,收率为73.5%。
DEI-MS,分子式C30H17N5S2,理论值511.09,测试值511.33。
实施例11化合物C36的制备
化合物C36的制备:在1L三口瓶中,氮气保护下,加入中间体7(3.2g,0.01mol),化合物A3(2.9g,0.011mol),随后加入100mL甲苯和50mL碳酸钠水溶液(2mol/L)。氮气保护下,回流搅拌加入Pd(PPh3)4(30mg,0.05mmol),回流反应12小时。反应完毕后向体系加入100mL水,分液。有机相脱溶剂后得到粗品,过硅胶柱,得到3.3g目标产物,纯度99.55%,收率为65.9%。
DEI-MS,分子式C28H15N5OS2,理论值501.07,测试值501.61。
实施例12化合物C39的制备
化合物C39的制备:在1L三口瓶中,氮气保护下,加入中间体7(3.2g,0.01mol),化合物A7(3.2g,0.011mol),随后加入100mL甲苯和50mL碳酸钠水溶液(2mol/L)。氮气保护下,回流搅拌加入Pd(PPh3)4(30mg,0.05mmol),回流反应12小时。反应完毕后向体系加入100mL水,分液。有机相脱溶剂后得到粗品,过硅胶柱,得到3.1g目标产物,纯度99.68%,收率为63.8%。
DEI-MS,分子式C28H15N5S2,理论值485.08,测试值485.51。
实施例13化合物C40的制备
化合物C40的制备:在1L三口瓶中,氮气保护下,加入中间体7(3.2g,0.01mol),化合物A6(3.3g,0.011mol),随后加入100mL甲苯和50mL碳酸钠水溶液(2mol/L)。氮气保护下,回流搅拌加入Pd(PPh3)4(30mg,0.05mmol),回流反应12小时。反应完毕后向体系加入100mL水,分液。有机相脱溶剂后得到粗品,过硅胶柱,得到3.0g目标产物,纯度99.67%,收率为59.7%。
DEI-MS,分子式C28H15N5OS2,理论值501.07,测试值501.44。
按照化合物样品制备的实施例1-13中所述的方法制备的有机光电材料(化合物C01-C41,相关化合物MS数据如下表1。
表1
从上述表1中的数据可以得知,本发明已经成功获得了所提供的有机光电材料,即式Ⅰ所示的有机光电材料。
实施例二制备有机电致发光器件(以下可简称为器件)
在下述制备有机电致发光器件的实施例中,所用到的试剂材料如下所示:
阳极:氧化铟锡(简称ITO)导电玻璃,空穴传输材料:NPB,
发光材料:mCP,电子传输材料:TPBI,电子注入材料:LiF,其中,NPB、mCP以及TPBI的结构式在前述中提到,在此不再赘述。
器件实施例1的制备
有机电致发光器件均按照下述方法进行制备:
a)清洗阳极:分别用去离子水、丙酮、乙醇超声清洗ITO导电玻璃,在上述溶剂中各超声清洗30分钟,然后在等离子体清洗器中处理5分钟;
b)在步骤a)中获得的阳极上真空蒸镀空穴传输材料NPB,获得空穴传输层,厚度为50nm;
c)在步骤b)中获得的空穴传输层上,真空蒸镀包括前述实施例1中制备得到的化合物C01发光材料,获得发光层,发光层的厚度为30nm,其中,化合物:mCP=1:10(W/W);
d)在步骤c)中获得的发光层上,真空蒸镀电子传输材料TPBI,获得电子传输层,电子传输层的厚度为30nm;
e)在步骤d)中获得的电子传输层上,真空蒸镀电子注入材料LiF,获得电子注入层,电子注入层的厚度为1nm;
f)在步骤e)中获得的电子注入层上,真空蒸镀阴极Al,阴极的厚度为100nm,获得有机电致发光器件。
在上述制备有机电致发光器件中,真空蒸镀时,压力小于1.0×10-3Pa。
由上述制备过程制备得到的有机电致发光器件,如图1中所示,包括阳极101、空穴传输层102、发光层103、电子传输层104、电子注入层105、阴极106,其中,空穴传输层102、发光层103、电子传输层104、电子注入层105均位于阴极层106和阳极101之间,在阳极101上依次叠置有空穴传输层102、发光层103、电子传输层104和电子注入层105。
器件实施例2-41的制备
本实施例与器件实施例1的不同之处在于:制备的有机电致发光器件的发光层使用本发明所提供的化合物C02-C41为材料。
器件对比例1
器件比较例1和器件实施例1不同的是:有机电致发光器件的发光层仅以mCP作为发光层材料,无本发明提供的有机光电材料。
选用日本拓普康公司SR3型分光辐射度计对上述实施例制备得到的有机电致发光器件进行下述测试,得到各个有机电致发光器件中的启亮电压、最大电流效率和光谱颜色。
在上述试验例中,检测各有机电致发光器件所得到的启亮电压、最大电流效率和光谱颜色如下表2中所示。
表2
由上述表2的结果,可以得知,本发明所提供的有机光电材料可应用于有机电致发光器件中。此外,通过由实施例中制备得到的器件的检测结果,还可以得知,本发明提供的有机光电材料使得有机电致发光器件获得优异的表现,本发明所提供的有机光电材料作为有机电致发光器件的发光材料使用,使得器件具有较大的最大电流效率,且使得器件具有较低的的启亮电压,同时,器件可以发出不同颜色的可见光,且光谱覆盖范围较广。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!