一种土茯苓精制多糖及其制备方法与流程
本发明涉及药学的技术领域,尤其是一种土茯苓精制多糖及其制备方法。
背景技术:
土茯苓(rhizomasmilacisglabrae)为百合科植物光叶菝葜的干燥根茎。其味甘、淡,性平,有解毒,除湿,通利关节的功效,主要用于治疗湿热淋浊,带下,瘰疬,疥藓,梅毒及汞中毒引起的肢体拘挛,筋骨疼痛(杨春昆.土茯苓临床应用研究进展[j].亚太传统医药,2017,13(17):77-78;国家药典委员会.中华人民共和国药典(2015版)一部[m].中国科技医药出版社,2015:8)。土茯苓多糖为土茯苓重要活性成分之一,其药理作用广泛,具有抗氧化、抗肿瘤、免疫调节、抗炎等生物活性,有很好的药用价值。这些数据都是关于粗多糖的,产业化价值有限。本课题组发现多糖是土茯苓主要成分之一,对其开展分离纯化、结构解析和生物活性评价具有十分重要的科学意义。
技术实现要素:
本发明要解决的技术问题是:为了解决上述背景技术中存在的问题,提供一种土茯苓精制多糖及其制备方法,制得的土茯苓精制多糖为均一多糖,得率高,具有抗氧化和神经保护活性,可以清除dph自由基、abst+,还原力测定强,可用于制备抗氧化剂和神经保护制剂。
本发明解决其技术问题所采用的技术方案是:一种土茯苓精制多糖,所述土茯苓精制多糖由半乳糖、甘露糖、阿拉伯糖和葡萄糖组成;所述土茯苓精制多糖的分子量为2.16×105da。
一种土茯苓精制多糖的制备方法,包括如下步骤:s1、将干燥土茯苓饮片,用5~8倍量乙醇加热冷凝回流2~3次进行脱脂;s2、将脱脂后的药渣用5~8倍量水提3次,温度为80~100℃,每次2h,将水提液减压浓缩后,加入无水乙醇至含醇量达80~85%,静置24h,弃去上清液,收集沉淀,得到土茯苓粗多糖;s3、将土茯苓粗多糖复溶后加胃蛋白酶至其含量为0.1~0.4%,于37℃下恒温水浴12h,去除沉淀,重复3次,收集上层溶液,浓缩,加入sevage试剂,剧烈振荡30min,静置12h,收集上层多糖溶液,重复至紫外扫描无蛋白特征吸收峰时止;s4、将s3中上层多糖溶液浓缩后加4~6倍量95%~100%乙醇,静置24h,收集沉淀,重复3次后进行冷冻干燥,得土茯苓粗多糖粉末;s5、将s4中获得的土茯苓粗多糖粉末用蒸馏水完全溶解后经deaefastflow离子层析柱分离,洗脱条件:流速为2.5ml/min,依次以水、0.05mol/l、0.1mol/l、0.2mol/l、0.3mol/l的氯化钠以及0.1mol/l的氢氧化钠进行洗脱;利用全自动收集器分梯度收集,每个溶液梯度洗脱3倍柱体积,收集30管,每管5ml,采用硫酸-苯酚法进行隔管跟踪检测;将收集到的土茯苓多糖溶液浓缩至一定体积后,用3500da分子量的透析带进行透析;装透析带的烧杯每天换水5次,透析一周后,将透析带内多糖液,离心,并将其进行冷冻干燥,得到初步纯化的土茯苓苓多糖;s6、将s5中收集的洗脱液浓缩后,用superdexg-200凝胶柱再分离,以蒸馏水按照流速0.2ml/min进行洗脱,利用全自动收集器收集,每管5ml,苯酚一硫酸法检测后,收集洗脱曲线中的主峰部分;s7、将s6中收集的洗脱曲线中的主峰部分的溶液浓缩后,用3500da的透析袋透析2d脱盐,最后将透析袋内的溶液进行浓缩冷冻干燥,得到土茯苓精制多糖.
进一步地限定,上述技术方案中,所述的s3sevage试剂中氯仿与正丁醇体积比为5∶1。
进一步地限定,上述技术方案中,将土茯苓精制多糖应用在制备抗氧化产品或在制备神经保护活性产品中。
进一步地限定,上述技术方案中,将羟丙甲纤维素、羧甲淀粉钠、微晶纤维素、乳糖、药用淀粉和硬脂镁与土茯苓精制多糖混合搅拌制成片剂。
进一步地限定,上述技术方案中,将微晶纤维素和药用淀粉与土茯苓精制多糖混合搅排制成片剂。
进一步地限定,上述技术方案中,将微晶纤维素、药用淀粉与土茯苓精制多糖混合搅拌,填入硬胶囊中,制成胶囊剂。
本发明的有益效果是:本发明提出的一种土茯苓精制多糖及其制备方法,制得的土茯苓精制多糖为均一多糖,得率高,具有抗氧化和神经保护活性,可以清除dph自由基、abst+,还原力测定强,可用于制备抗氧化剂和神经保护制剂。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本申请中记载的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为土茯苓粗多糖组分过deaefastflow离子层析柱的洗脱曲线图;
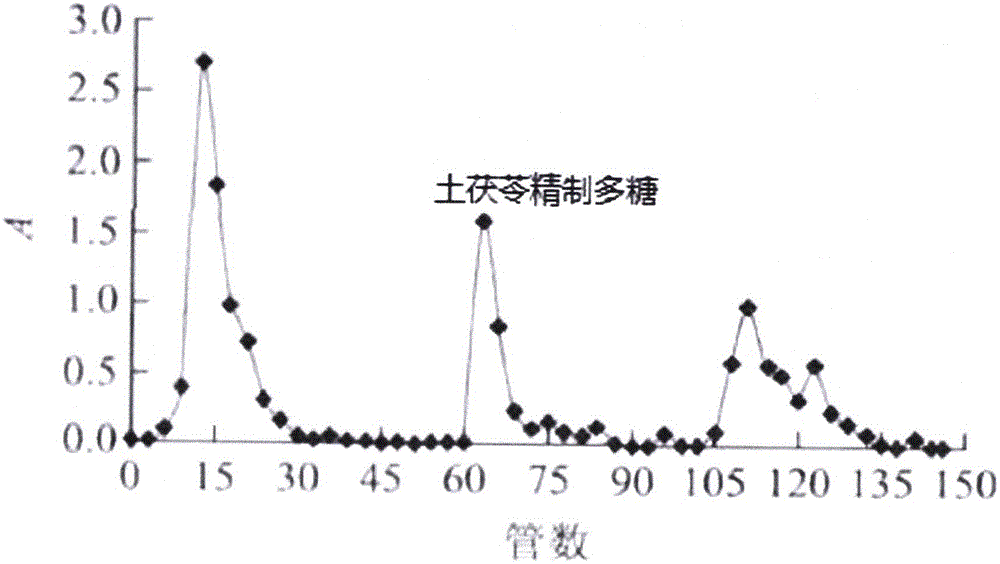
图2为土茯苓精制多糖的hplc色谱图;
图3为标准单糖gc-ms总离子流色谱图;
图4土茯苓精制多糖gc-ms.总离子流色谱图;
图5为土茯苓精制多糖的1r图;
图6为土茯苓精制多糖的1h谱图;
图7为土茯苓精制多糖的13c谱图。
具体实施方式
为了使本发明所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
见图1~7所示的是一种土茯苓精制多糖,所述土茯苓精制多糖由半乳糖、甘露糖、阿拉伯糖和葡萄糖组成;所述土茯苓精制多糖的分子量为2.16×105da。
一种土茯苓精制多糖的制备方法,包括如下步骤:s1、将干燥土茯苓饮片,用5~8倍量乙醇加热冷凝回流2~3次进行脱脂;s2、将脱脂后的药渣用5~8倍量水提3次,温度为80~100℃,每次2h,将水提液减压浓缩后,加入无水乙醇至含醇量达80~85%,静置24h,弃去上清液,收集沉淀,得到土茯苓粗多糖;s3、将土茯苓粗多糖复溶后加胃蛋白酶至其含量为0.1~0.4%,于37℃下恒温水浴12h,去除沉淀,重复3次,收集上层溶液,浓缩,加入sevage试剂,剧烈振荡30min,静置12h,收集上层多糖溶液,重复至紫外扫描无蛋白特征吸收峰时止;s4、将s3中上层多糖溶液浓缩后加4~6倍量95%~100%乙醇,静置24h,收集沉淀,重复3次后进行冷冻干燥,得土茯苓粗多糖粉末;s5、将s4中获得的土茯苓粗多糖粉末用蒸馏水完全溶解后经deaefastflow离子层析柱分离,洗脱条件:流速为2.5ml/min,依次以水、0.05mol/l、0.1mol/l、0.2mol/l、0.3mol/l的氯化钠以及0.1mol/l的氢氧化钠进行洗脱;利用全自动收集器分梯度收集,每个溶液梯度洗脱3倍柱体积,收集30管,每管5ml,采用硫酸-苯酚法进行隔管跟踪检测;将收集到的土茯苓多糖溶液浓缩至一定体积后,用3500da分子量的透析带进行透析;装透析带的烧杯每天换水5次,透析一周后,将透析带内多糖液,离心,并将其进行冷冻干燥,得到初步纯化的土茯苓苓多糖;s6、将s5中收集的洗脱液浓缩后,用superdexg-200凝胶柱再分离,以蒸馏水按照流速0.2ml/min进行洗脱,利用全自动收集器收集,每管5ml,苯酚一硫酸法检测后,收集洗脱曲线中的主峰部分;s7、将s6中收集的洗脱曲线中的主峰部分的溶液浓缩后,用3500da的透析袋透析2d脱盐,最后将透析袋内的溶液进行浓缩冷冻干燥,得到土茯苓精制多糖.
其中,s3sevage试剂中氯仿与正丁醇体积比为5∶1。
土茯苓粗多糖的提取:
取土茯苓药材于圆底烧瓶中,按1∶10(g∶ml)的料液比加入乙醇,加热冷凝回流2h,进行第二次脱脂时料液比为1∶8(g∶ml),回流2h。将脱脂后的药渣晾晒,挥发掉残留的乙醇,按1∶10(g∶ml)的料液比向土茯苓药渣中加蒸馏水,提取3次,2h/次,温度为90℃,提取结束后抽滤,合并3次的水提液。对提取液进行减压浓缩,加入4倍量的乙醇,边加边搅拌,于4℃下静置24h后抽滤,弃去上清液,收集沉淀,得到粗多糖。
土茯苓精制多糖的纯化:
称取土茯苓粗多糖用蒸馏水溶解,经deaefastflow离子层析柱分离,洗脱条件:流速为2.5ml/min,依次以水、0.05mol/l、0.1mol/l、0.2mol/l、0.3mol/l的氯化钠以及0.1mol/l的氢氧化钠进行洗脱。利用全自动收集器分梯度收集,每个溶液梯度洗脱3倍柱体积,收集30管,每管5ml,采用硫酸-苯酚法进行隔管跟踪检测。将收集到的土茯苓多糖溶液浓缩至一定体积后,用3500da分子量的透析带进行透析(透析带须进行预处理)。装透析带的烧杯每天换水5次,透析一周后,将透析带内多糖液,离心,并将其进行冷冻干燥,得到初步纯化的土茯苓苓多糖,然后再用蒸馏水充分溶解,采用superdexg-200凝胶柱再分离,以蒸馏水按照流速0.2ml/min进行洗脱,利用全自动收集器收集,每管5ml,苯酚一硫酸法检测后,收集洗脱曲线中的主峰部分。收集到的溶液浓缩后用截留分子量为3500da的透析袋透析2d来脱盐,收集袋内溶液进行浓缩,之后冷冻干燥,得到土茯苓精制多糖。
土茯苓均多糖的纯度鉴定:
高效液相色谱条件:agilent1200高效液相色谐仪,色谱柱为g-5000pwxl和tskg-3000pwxl串联,流动相为0.02mol/lkh2p04溶液,流速为0.6ml/min,流动相为0.2mol/l的nano3溶液,流速为0.6ml/min,柱温35℃,检测器为waters2414示差折光检测器。将步骤3获得的土茯苓多糖用适量水溶解,进样为20μl,结果色谱图中呈单一对称峰,说明该组分为均一多糖,经测定纯度为98.86%,得率为3.08%。
土茯苓精制多糖分子量的确定:
取1mg分子量为50k、80k、150k、270k、410k和670kda的普鲁兰多糖分别溶于1ml水中,色谱条件同上,用agilent1200高效液相色谱仪分析,记录保留时间,以分子的对数(logm)为纵坐标,保留时间(t)为横坐标,得标准曲线y=7.965+0.986x,将土茯苓精制多糖出峰时间代入曲线方程,得土茯苓精制多糖的分子量为2.16×105da。
土茯苓精制多糖的结构表征:
1.多糖组分分析:
先进行酸水解,然后乙酰化进行衍生,再进行气相gc分析。酸水解的方法是:称取10mg土茯苓精制多糖样品,分别置于安培瓶中,加入浓度为2mol/l的4ml三氟乙酸,氮气吹跑瓶内空气,用酒精喷灯封管。110℃水解6小时后,旋蒸干样品,加入2ml的甲醇溶解,蒸干,反复3次,尽量去除样品中的三氟乙酸。加入1ml的甲醇将样品转移至血清瓶内,氮气吹干,然后按照上述方法衍生化后,进行gc分析,其结果根据单糖标准品的保留时间进行分析,并通过出峰的面积比计算出单糖摩尔百分比。结果显示,同单糖标准品的gc图谱中的保留时间对照,结果表明土茯苓精制多糖由半乳糖、甘露糖、阿拉伯糖和葡萄糖组成,根据内标法计算出四种单糖的摩尔比是4.46∶1.00∶2.22∶30.88。甲基化研究方法:称取土茯苓多糖样品20mg(样品保持充分的干燥)置于带塞试管中,用6ml的二甲基亚砜dmso试剂,并用氮气封口,进行加热,磁力搅拌混匀,加入氢氧化钠(6ml的dmso含有240mg的氢氧化钠)形成氢氧化钠悬浊液,过夜。隔日向试管中加入3.6ml的碘甲烷ch3i,搅拌8min,用氮气吹走ch3i,再次甲基化,如此重复3次后,加入6ml的蒸馏水中止反应。流水和去离子水各透析24h后,用氯仿chcl3萃取3次后,用无水亚硫酸钠干燥24h,然后氮气吹干,溶液剩下约1ml。采用三氟乙酸水解的方法对其进行水解,再加入70mg的硼氢化钠nabh4搅拌24h,再加强酸性阳离子交换树脂搅拌混匀10min,抽滤,收集滤液加甲醇,氮气吹干,乙酰化方式进行衍生化,再进行gc-ms分析。
2.土茯苓精制多糖ir分析
取2mg土茯苓精制多糖样品与200mg干燥的kbr混合研磨、压片,以nicolet5700傅里叶变换红外光谱仪在400~4000cm-1范围内进行扫描,初步判定多糖基团结构。从ir图中可以看出,土茯苓精制多糖呈现典型的多糖吸收特征,其中在3386.85cm-1处有强且宽的吸收峰,这是由于糖分子内或分子间氢键的o-h伸缩振动引起的,在2929.06cm-1出现甲基的c-h键伸缩振动峰,在1639.45cm-1处出现少量羰基吸收峰。在1373.00、1247.83cm-1处存在c-h变角振动的弱吸收峰。在1152.82、1025.50、931.38cm-1的3个吸收峰为吡喃环的特征吸收峰,其中1152.82cm-1和1025.50cm-1为吡喃环的醚键(c-o-c)伸缩振动和羟基(c-o-h)的吸收峰,在931.38cm-1的吸收峰是α型吡喃糖的特征吸收峰。
3.土茯苓精制多糖甲基化分析:
甲基化反应:分别取10mg干燥的土茯苓精制多糖和土茯苓精制多糖糖酸还原产物加入到25ml的圆底烧瓶中,加2ml无水dmso溶液超声30min溶解,加30mg干燥的naoh粉末超声反应3h,冰浴下沿壁级慢滴加1mlch3i,避光超声反应2h,加3ml水结束反应,按1∶1加chci3萃取,收集有机相,重复3次,干燥后重复甲基化至ir扫描图谱中没有羟基峰。整个反应过均在20℃,n2保护下进行。后续反应:向甲基化后的样品中加4ml2mol/l的tfa,于110℃下水解5h,水解后4ml甲醇旋干除去多余的tfa,重复3次,再加0.5ml.水溶解;取0.2ml.水解液,加30mgnabh4室温下反应3h,反应过程中间歇振摇,结束后滴加25%冰乙酸酸至无气泡产生,加2ml甲醇旋干,重复3次;向还原产物中加1.5ml乙酸,于100℃下反应1h后,加2ml甲苯旋干,重复3次以除去多余的乙酸酐;向乙酰化产物中各加2ml氯仿和水,振荡,静置,收集下层有机相,用lml水洗涤3次,再加少量无水硫酸钠除水,待gc-ms分析。
gc-ms色谱条件:agilent6890-5973n型气相色谱-质谱联用仪,色谱柱:hp-5ms毛细管柱(30m×250pm×0.25umd);载气:he;加热器温度:250℃:程程序升温:初始温度140℃/min升至200℃,保持5min,再以8℃/min升至240℃:分流比:50∶1;进样量:5μl。
表1土茯苓精制多糖甲基化分析数据
见表1,土茯苓精制多糖甲基化的结果表明,土茯苓精制多糖主要是→4)glcp(1→、→6)glcp(1→、→4galp(1→组成,glcp残基是土茯苓多糖的主要单元,以1→4、1→6、1→4,6和1→连接方式存在,galp残基以1→4连接方式存在。
4.土茯苓精制多糖nmr分析
取30mg干燥的土茯苓精制多糖,溶于0.5mld20中,进行nmr分析。1h-nmr谱中,α型吡喃糖的异头碳上质子的化学位移通常高于5.0,而β型则低于5.0。在土茯苓精制多糖的1h-nmr谱中,异头碳的强信号为4.7~4.9和3.6~3.4,可见土茯苓精制多糖的糖以β构型为主。13c-nmr谱中,糖残基的异头碳在13c-nmr上的共振信号在δ90~110范围内,可以通过峰的个数来确定糖残基的数目和相对含量。α型糖苷异头碳的共振信号在δ95~101范围,而β型糖苷异头碳的共振信号在δ101~110范围。土茯苓精制多糖异头碳区域的的化学位移值处于δ106.22ppm,可以判断葡聚糖的糖残基为吡喃环型,糖苷键的连接方式为β型,这与1h-nmr的推测相一致。
5.土茯苓精制多糖的抗氧化活性实验
5.1清除dpph自由基活性测定
参照golluecke等的方法测定,取2ml不同质量浓度的样品溶液(12.5,25,50,100μg·ml-1),加入2mldpph溶液(100μg·ml-1),充分混匀后,避光反应30min。于517nm处测定反应体系的吸光度值(am)。同时设定溶剂空白组(an,dpph溶液用等体积甲醇代替)和样品空白组(ao,样品溶液用等体积甲醇代替)。vc作为阳性对照组。实验平行操作3次,根据下列公式计算提取液对dpph的清除率。
表2土茯苓精制多糖清除dpph自由基活性
与维生素c组比较,*p<0.05,**p<0.01
5.2清除abts+活性测定
参照robertare等的方法测定,取0.4ml不同质量浓度的样品溶液(12.5,25,50,100μg·ml-1),加入abts+溶液4ml,反应6min,于734nm处测定其吸光度ai。同时设定溶剂空白组(aj,abts+溶液用等体积甲醇代替),样品空白组(ah,样品溶液用等体积甲醇代替)和阳性对照组。实验平行操作3次,根据下列公式计算提取液对abts+的清除率:
表3土茯苓精制多糖清除abst+活性
与维生素c组比较,*p<0.05,**p<0.01
5.3还原力测定
参照mazord等的方法测定,取0.8ml不同质量浓度的样品溶液(12.5,25,50,100μg·ml-1),加入2ml磷酸盐缓冲液(ph=6.6)和2ml1%铁氢化钾,50℃水浴20min,加2ml10%三氯乙酸,3000rpm,离心10min,取2ml上清加2ml去离子水,0.4ml0.1%三氯化铁,反应5min,于700nm处测吸光度值,以vc为阳性对照,实验平行操作3次,测得的吸光度值越大表示还原能力越强。
表4土茯苓精制多糖还原力测定
与维生素c组比较,*p<0.05,**p<0.01
结果:土茯苓精制多糖清除dpph自由基和abts+的ic50值分别为7.04±1.22μg·ml-1和29.57±5.46μg·ml-1,还原力ec50值为为74.14±19.33μg·ml-1,和阳性药维生素c相当,这说明土茯苓精制多糖具有较强的抗氧化活性。6.土茯苓精制多糖的神经保护活性实验。
将乳鼠颈椎脱臼处死后,在无菌条件下取完整鼠脑部,解剖镜下剥离脑膜和血管等纤维成分,0.25%胰蛋白酶消化过滤,1500r/min离心5分钟,无菌微孔滤膜过滤后10%胎牛血清培养。将其接种于25cm2的塑料培养瓶中,密度为每瓶2×107个细胞,放置5%c02、37℃、饱和湿度的二氧化碳培养箱中静置原代培养。48h后换液,7~10天长满备用。将大鼠原代神经胶质细胞分为6组,每组6份,1组为空白对照组,2组为lps(10μg/l)组,3、4、5组分别为lps(10μg/l)+土茯苓精制多糖处理组,土茯苓精制多糖的浓度分别为低剂量、中剂量和高剂量,实验重复6次。空白对照组不加药,lps模型组均加入lps作用2h,3个给药组培养皿加入干燥的土茯苓精制多糖作用24h。采用mtt实验检测细胞的增殖能力,采用硝酸还原酶法检测土茯苓精制多糖对一氧化氮的影响,运用酶联免疫吸附法(elisa)测定培养上清中il-1β、il-8、tnf-α水平,通过实时逆转录聚合酶链反应(real-timert-pcr)测定细胞中诱导型一氧化氮合酶(induciblenitricoxidesynthase,inos)mrna表达。结果显示,lps能够激活神经胶质细胞,不同浓度的土茯苓精制多糖在不影响细胞存活率的情况下,可以显著降低细胞培养上清液中no、il-1β、il-8、tnf-α水平,细胞内inosmrna表达也明显减少(p<0.05),土茯苓精制多糖神经保护的机制可能与抑制胶质细胞炎症有关。
表5土茯苓精制多糖对lps诱导神经胶质细胞存活率的影响
与空白对照组比较,*p<0.05,**p<0.01;与lps组比较,δp<0.05,δδp<0.01
表6土茯苓精制多糖对lps诱导神经胶质细胞inosmrna表达的影响
与空白对照组比较,*p<0.05,**p<0.01;与lps组比较,δp<0.05,δδp<0.01
表7土茯苓精制多糖对上清液中n0、tnf-α、il-1β、il-8水平的影响
与空白对照组比较,*p<0.05,**p<0.01;与lps组比较,δp<0.05,δδp<0.01
实施例一:
取土茯苓精制多糖2g、羟丙甲纤维素4g、羧甲淀粉钠10g,微晶纤维素10g、乳糖115g、药用淀粉50g、硬脂酸铁1g,将主药与辅料充分混匀后投入速搅拌机中,喷雾加水适量,整粒,水分控制在3~4%,然后月压片,制成1000片,包薄膜衣。每片含主药成分0.2mg。该片剂为抗氧化产品或者神经保护产品。
实施例二:
土茯苓精制多糖3g、羧甲淀粉钠15g、微晶纤维素15g、药用淀50g、硬脂酸镁2g,将主药与辅料充分混匀后投入高速搅拌机中,喷雾加水适量,整粒,水控制在3~4%,然后压片,制成1000片。每片含主药成分0.3mg。该片剂为抗化产品或者神经保护产品。
实施例三:
取土茯苓精制多糖2g、微晶纤维素25g、药用淀粉70g,将药用淀先干燥,过120目筛,再与土茯苓精制多糖、微晶纤维素混合,过两次120日筛,填入硬胶囊制成1000粒本发明胶囊。每粒硬胶囊含主药成分0.2mg,该胶囊为抗氧化产品或者神经保护产品。
以上所述的,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!