一种基于解旋酶和切刻酶的等温扩增核酸检测方法及试剂盒与流程
本发明涉及分子生物学核酸检测
技术领域:
,具体地,涉及一种基于解旋酶和切刻酶的等温扩增核酸检测方法及试剂盒。
背景技术:
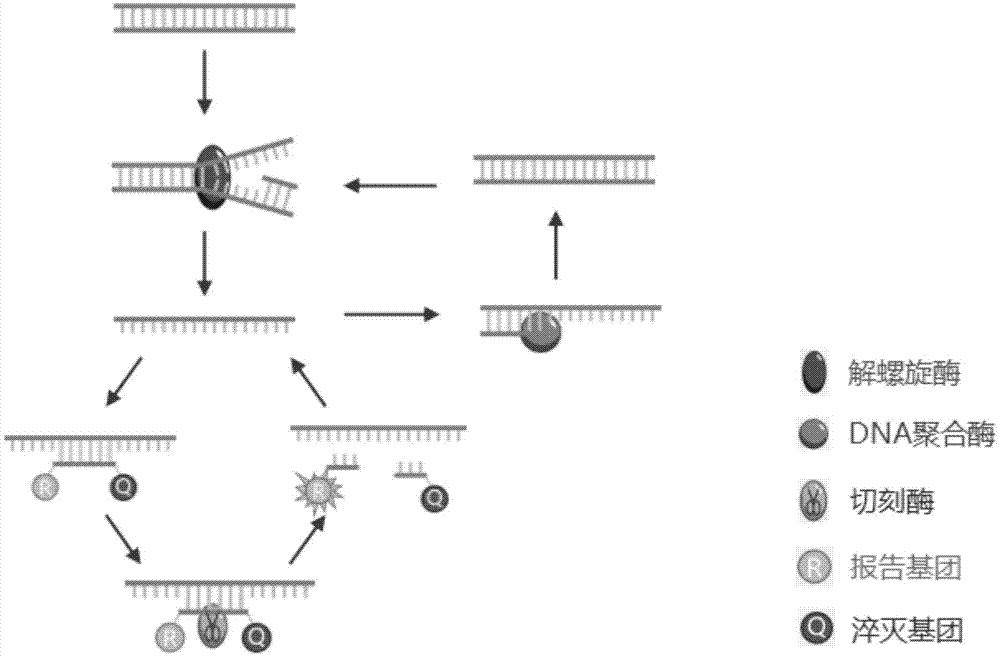
核酸检测技术已广泛应用于食品检测、环境检测及疾病的预防和控制等方面。其中聚合酶链式反应(polymerasechainreaction,pcr)的高灵敏度使其成为目前使用最广泛的dna扩增方法,然而现有的常规pcr技术需要反复的热变性以解开dna双链,在应用上依赖于高质量的热循环,且扩增效果易受多因素影响、反应时间长等。等温扩增技术是核酸体外扩增技术,其反应过程始终维持在恒定的温度下,通过添加不同活性的酶和各自特异性引物来达到快速核酸扩增的目的。等温扩增技术对仪器要求大大简化,反应时间相比pcr技术大大缩短,且特异性高、成本低廉,更能满足快速检测的需求。常用的核酸等温扩增技术有环介导等温扩增技术(lamp)、链置换扩增(sda)、依赖解旋酶的等温扩增技术(had)、滚环扩增技术(rca)等。环介导的恒温扩增(lamp)法虽然扩增效率高,特异性强,但是该方法对引物设计的要求特别高,引物设计复杂,需要四条能够识别靶序列六个特异区域的引物,在同一反应管中进行多重检测的难度比较大;lamp所识别的靶位序列长度不得过大,无法对长片段进行扩增;因具备较高的灵敏度,需对操作要求严格分区,否则极易受到污染产生假阳性结果等。滚环扩增(rollingcircleamplification,rca)是于恒定温度下以单链环状dna为模板,在有强链置换活性的phi29dna聚合酶作用下,通过引物与模板环退火而进行的滚环式dna合成,rca极高的扩增效率使其可成为信号放大的手段,但是在rca反应过程中未成环的锁式探针和未结合探针的模板dna或者rna可能产生一些背景信号。链置换等温扩增(sda)方法是基于在靶dna两端带有被化学修饰的限制性核酸内切酶识别序列,核酸内切酶在其识别位点将链dna打开缺口,dna聚合酶延伸缺口3’端并替换下一条dna链,被置换下来的dna单链可与引物结合并被dna聚合酶延伸成双链,从而实现扩增。但该技术对于引物和靶序列结合形成5’端有特殊要求。sda在等温扩增之前需要一个加热变性打开双链的步骤,由于klenowfragmentexo-无热稳定性,必须在靶dna变性后才能加入到体系中,容易引起污染。为了引入切刻内切酶识别的特异序列,通常也需要3条或3条以上引物。另外,也有依赖解旋酶或重组酶的方法进行等温扩增,利用解旋酶或重组酶将dna双链打开,进入扩增循环,这些方法都需要对靶标序列进行指数级扩增,以便检测,这就和其他pcr扩增相同,都会产生大量pcr扩增产物,在操作疏忽或者没有高级通风设备的实验室或者没有功能区分割的小型实验室中很容易造成核酸气溶胶的扩散,从而导致严重污染。这点也限制了等温扩增在现场快速检测(poct)中的应用。因此,目前需要开发一种新的适用于现场快速检测核酸的方法,以减少操作过程中产生的污染进而影响核酸检测准确性和结果。技术实现要素:本发明的目的是为了克服现有技术的上述不足,提供一种基于解旋酶和切刻酶的等温扩增核酸检测方法,该方法可有效解决常规扩增大量核酸产物引起的实验室污染问题,原理简单,操作方便,特异性强,灵敏度高。本发明的另一目的在于提供一种基于解旋酶和切刻酶的等温扩增核酸检测试剂盒。为了实现上述目的,本发明是通过以下方案予以实现的:一种基于解旋酶和切刻酶的等温扩增核酸检测方法,包括如下步骤:s1.根据目标核酸序列设计特异性扩增引物和带有切刻酶识别位点的taqman探针;s2.将样本核酸经酶处理得到单链目标核酸模板;s3.将s1所得taqman探针与s2所得单链目标核酸模板特异性结合,得到特异性结合产物,使用切刻酶酶切特异性结合产物,并实时检测荧光信号。本发明通过设计带有切刻酶识别位点的特异性taqman探针,探针与目标单链模板结合后可被切刻酶切割,探针被切断后因为tm值降低,就会从靶位点上脱落,荧光基团和淬灭基团分离,产生荧光信号。而新的探针会继续特异性结合到目标单链模板,再次被切刻酶切割,产生荧光,信号持续进行,产生大量荧光信号。本发明将解旋酶与切刻酶联合使用,设计带有切刻酶切割位点的特异性taqman探针,不需要扩增出大量核酸产物,只需要产生适量单链模板,然后用切刻酶不断切割和靶位点结合的探针即可进行检测,可以避免等温扩增很容易造成实验室污染的缺点,操作简便、快速。当样本为双链核酸时,步骤s2中双链核酸样本经酶处理得到用于扩增和检测的单链目标核酸模板。优选地,步骤s2所述酶为解螺旋酶、链置换酶、重组酶中的一种或多种。更优选地,步骤s2所述酶为uvrd解旋酶和链置换dna聚合酶。当样本为单链rna时,优选地,步骤s2中rna样本在逆转录酶的作用下,形成rna-dna杂交双链,再采用rnaseh分解rna-dna杂交体系中的rna链,形成扩增和检测的单链目标核酸模板。即此时步骤s2中所述酶为逆转录酶和rnaseh。优选地,当单链目标核酸模板过少时,通过向反应体系中加入特异性扩增引物和聚合酶,使单链目标核酸模板在特异性扩增引物、聚合酶和步骤s2所述酶交替作用下不断扩增。通过步骤s2所述酶处理双链核酸产生目标单链片段,目标单链片段在特异性引物和聚合酶的作用下进一步合成双链核酸,合成的双链核酸又不断在步骤s2所述酶的作用下产生目标单链片段;循环往复,不断产生单链目标片段。目标单链模板的生成只需在恒温下通过一对特异性引物和酶进行作用即可得到,技术原理简单,操作方便,特异性强。优选地,所述步骤s3的具体过程为:taqman探针与单链目标核酸模板特异性结合,切刻酶作用于探针的切刻酶识别位点,探针被切断后从单链核酸模板上脱落,并产生荧光信号;新的探针再次与单链核酸模板结合,再次被切刻酶切断,从单链模板脱落,产生荧光信号;探针的结合-切断-脱落过程重复进行,不断产生荧光信号。通过abi7500实时荧光定量pcr仪对产生的荧光信号进行实时监测,每分钟检测一次荧光信号。优选地,所述检测的条件为反应温度50~60℃,检测时间90min。本发明还请求保护一种基于解旋酶和切刻酶的等温扩增核酸检测试剂盒,包括以下终浓度的各组分:8~12μm特异性扩增引物、8~12μmtaqman探针、180~220ng/μl酶、4~6μg/μlt4基因32蛋白、5~10u/μl聚合酶、8~12u/μl切刻酶、8~12mmdntp。优选地,所述等温扩增核酸检测试剂盒还包括5×缓冲液;所述5×缓冲液包括以下终浓度的各组分:25mmtris-hcl、ph7.8,125mmnacl,50mm(nh4)2so4,250mmmgcl2,2.5mg/mlbsa,1m甜菜碱,0.5%tween20。优选地,所述聚合酶为大肠杆菌dna聚合酶i的klenow片段、t7dna聚合酶、bst聚合酶中的任一种。更优选地,所述聚合酶为bst聚合酶。优选地,所述切刻酶为nb.bbvci、nb.bsmi、nb.bsrdi、nb.bsssi、nb.btsi、nt.alwi、nt.bbvci、nt.bsmai、nt.bspqi、nt.bstnbi、nt.cvipii中的任一种。更优选地,所述切刻酶为nt.bstnbi。与现有技术相比,本发明具有以下有益效果:(1)本发明所述检测方法不需要对靶序列进行大量扩增,只要得到适量单链模板,通过taqman探针被不断切割即可产生大量的荧光信号进行检测,避免了常规荧光检测方法中的背景片段干扰问题,极大降低了实验室污染产生的可能性,能够在条件普通的实验室进行实验操作。(2)本发明所述检测试剂盒原理简单易懂,操作便捷,只需将提取的样本核酸与该试剂盒反应体系混合,55℃下即可自动进行反应,实时检测荧光信号,快速灵敏,可用于现场快速检测。附图说明图1为本发明所述检测方法检测dna的基本原理图。图2为本发明所述检测方法检测rna的基本原理图。图3为本发明实施例1中人基因组gapdh基因的检测结果。图4为本发明实施例2中艰难梭菌tcdb产毒基因的检测结果。图5为本发明实施例3中病原菌检测特异性分析结果。图6为本发明实施例4中人源gapdh基因的mrna检测结果。具体实施方式下面结合说明书附图及具体实施例对本发明作出进一步地详细阐述,所述实施例只用于解释本发明,并非用于限定本发明的范围。下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。实施例1人基因组gapdh基因检测1、检测试剂盒制备(1)特异性引物设计针对人类基因组gapdh基因,综合考虑解链温度、gc含量、引物长度、引物结构、引物二聚体等多个参数,设计特异性引物对,具体如下所示。扩增片段(seqidno:1):cgggtgatgcttttcctagattattctctggtaaatcaaagaagtgggtttatggaggtcctcttgtgtcccctccccgcagaggtgtggtggctgtggcatggtgccaagccgggagaagctgagtcatgggtagttggaaaaggacatttccaccgcaaaatggcccctctg上游引物primerf(seqidno:2):5’-cgggtgatgcttttcctagatt-3’下游引物primerr(seqidno:3):5’-cagaggggccattttgcgg-3’(2)特异性taqman探针设计根据单链目标核酸模板设计特异性taqman探针,且探针序列中含有切刻酶识别位点。探针序列的5’端连接有荧光基团,3’端连接有淬灭基团。当探针处于正常长度时,激发光照射时荧光基团的发射光能够被淬灭基团吸收,此时检测不到荧光信号;当探针被切断后,因为tm值降低,就会从靶位点上脱落,荧光基团和淬灭基团分离,产生荧光信号。切刻酶的特点是在识别位点进行切割,但是只能切断双链中的一条链。即当探针特异性结合到单链目标核酸模板上后,形成双链,切刻酶识别位点切断探针,探针从单链目标核酸模板上脱离,产生荧光信号。探针序列probe(seqidno:4):5’-fam-ctgagtcatgggtagttggaaa-bhq-3’(3)反应体系配制每20μl的扩增反应体系中含有4μl5×缓冲液、2μl10mmdntp、1μl8u/μlbst聚合酶、0.5μl200ng/μluvrd解旋酶、0.5μl5μg/μlt4基因32蛋白(gp32)、0.6μl10u/μlnt.bstnbi、1.5μl10μmprimerf、1.5μl10μmprimerr、1μl10μmprobe、2μl样本dna、5.4μlddh2o。其中5×缓冲液由25mmtris-hcl(ph7.8)、125mmnacl、50mm(nh4)2so4,250mmmgcl2、2.5mg/mlbsa、1m甜菜碱、0.5%tween20组成。2、人基因组gapdh基因检测,具体步骤为:(1)收集培养的人源细胞hek293,按照常规方法提取基因组dna。(2)将基因组dna提取液稀释为不同浓度1μg/μl、100ng/μl、10ng/μl、1ng/μl、0.1ng/μl,分别取2μldna稀释液加入到20μl的扩增体系中,每个扩增反应中的dna浓度为100ng/μl、10ng/μl、1ng/μl、0.1ng/μl、0.01ng/μl,空白对照组加入2μlddh2o。(3)配制20μl的扩增反应体系,在振荡器上混匀,放入abi7500实时荧光定量pcr仪中,设置反应温度为55℃,反应时间为90分钟,每分钟检测一次荧光信号。检测结果如图3所示,结果显示本发明所述检测方法可以从人类基因组中检测gapdh基因,检测下限为0.1ng/μldna。实施例2艰难梭菌产毒基因tcdb检测1、检测试剂盒制备(1)特异性引物和探针设计针对检测靶标序列艰难梭菌产毒基因tcdb设计特异性引物和探针,具体如下所示。扩增片段(seqidno:5):cacaagtggtagaagaaaggattgaagaagctaaaagcttaacttctgactctattaattatataaagaatgaatttaaactaatagaatctatttctgatgcactatacgatttaaaacaacagaatgaattagaagagtctcattttatatcttttgaggatatatcggagactgatgaaggct上游引物primerf(seqidno:6):5’-cacaagtggtagaagaaaggattg-3’下游引物primerr(seqidno:7):5’-agccttcatcagtctccgata-3’探针序列probe(seqidno:8):5’-fam-aagagtctcattttatatctttt-bhq-3’(2)反应体系配制每20μl的扩增反应体系中含有4μl5×缓冲液、2μl10mmdntp、1μl8u/μlbst聚合酶、0.5μl200ng/μluvrd解旋酶、0.5μl5μg/μlt4基因32蛋白(gp32)、0.6μl10u/μlnt.bstnbi、1.5μl10μmprimerf、1.5μl10μmprimerr、1μl10μmprobe、2μl样本dna、5.4μlddh2o。其中5×缓冲液由25mmtris-hcl(ph7.8)、125mmnacl、50mm(nh4)2so4,250mmmgcl2、2.5mg/mlbsa、1m甜菜碱、0.5%tween20组成。2、艰难梭菌产毒基因tcdb检测,具体步骤为:(1)在厌氧箱中进行艰难梭菌培养,然后按照常规方法提取艰难梭菌的基因组dna,被检测艰难梭菌菌液的浓度分别为150cfu/ml、300cfu/ml、500cfu/ml、1500cfu/ml、5000cfu/ml。(2)将不同浓度的艰难梭菌基因组dna提取液分别取2μldna加入到20μl的扩增体系中,空白对照组加入2μlddh2o。(3)配制20μl的扩增反应体系,在振荡器上混匀,放入abi7500实时荧光定量pcr仪中,设置反应温度为55℃,反应时间为90分钟,每分钟检测一次荧光信号。检测结果如图4所示,检测下限为300cfu/ml,说明本发明所述方法可满足病原菌检测的基本需求。实施例3病原菌检测特异性分析采用实施例2所述试剂盒和检测方法,选择大肠杆菌、沙门氏菌、金黄色葡萄球菌、鲍氏志贺氏菌、痢疾志贺氏菌、双酶梭状芽胞杆菌、产气荚膜梭菌和不产毒的艰难梭菌为检测样本,检测艰难梭菌产毒基因tcdb,分析实施例2所述试剂盒和检测方法在病原菌检测方面的特异性,具体实验设置如表1所示。表1病原菌检测特异性分析实验设置实验组病原菌浓度1产毒艰难梭菌5000cfu/ml2产毒艰难梭菌500cfu/ml3大肠杆菌、沙门氏菌106cfu/ml4金黄色葡萄球菌、鲍氏志贺氏菌106cfu/ml5痢疾志贺氏菌、双酶梭状芽胞杆菌106cfu/ml6产气荚膜梭菌、不产毒的艰难梭菌106cfu/ml扩增反应体系配好后,在振荡器上混匀,放入abi7500实时荧光定量pcr仪中,设置反应温度为55℃,反应时间为90分钟,每分钟检测一次荧光信号。结果如图5所示,加入产毒艰难梭菌的检测管中,有强荧光信号,而加入其它细菌dna的管中基本无荧光信号,其中也包括不产毒的艰难梭菌dna,因为不产毒的艰难梭菌无tcdb基因,因此不会出现荧光信号。以上结果表明本发明所述检测方法具有良好的特异性,能够特异的对目的基因进行识别。实施例4rna检测1、检测试剂盒制备(1)特异性引物设计针对人源基因gapdh的mrna,设计特异性引物对和探针,具体如下所示。扩增片段(seqidno:9):acccattcattgaccttgattacatggtttacatgttccagtacgactccacccatggaaagtacaagggtgaggttaaggcagaaggcggcaaactggtcattgatggtcatgcaatcacagtctatagcgagagggacccagccaacattaagtggggtgatgcaggtgctacttatgttgtggagtctac上游引物primerf(seqidno:10):5’-acccattcattgaccttgat-3’下游引物primerr(seqidno:11):5’-gtagactccacaacataagt-3’探针序列probe(seqidno:12):5’-fam-tggagtcgtactggaacatgta-bhq-3’(2)扩增反应体系配制每20μl的扩增反应体系中含有4μl5×缓冲液、2μl10mmdntp、1μl8u/μlbst聚合酶、0.5μl200ng/μluvrd解旋酶、0.5μl5μg/μlt4基因32蛋白(gp32)、0.6μl10u/μlnt.bstnbi、1.5μl10u/μlamv逆转录酶、1μl5u/μlrnaseh、1.5μl10μmprimerf、1.5μl10μmprimerr、1μl10μmprobe、2μl样本dna、2.9μlddh2o。其中5×缓冲液由25mmtris-hcl(ph7.8)、125mmnacl、50mm(nh4)2so4,250mmmgcl2、2.5mg/mlbsa、1m甜菜碱、0.5%tween20组成。2、人源基因gapdh的mrna检测,具体步骤为:(1)收集培养的人源细胞hek293,按照常规方法提取hek293细胞的总rna。(2)将rna提取液稀释为不同浓度100ng/μl、10ng/μl、1ng/μl,总rna于70℃加热5min后置于冰上3min,分别取2μl加入到20μl的扩增体系中,每个扩增反应中的rna含量为10ng/μl、1ng/μl、0.1ng/μl,空白对照组加入2μlddh2o。(3)配制20μl的扩增反应体系,在振荡器上混匀,放入abi7500实时荧光定量pcr仪中,设置反应温度为52℃,反应时间为90分钟,每分钟检测一次荧光信号。检测结果如图6所示,结果显示本发明所述检测方法可以有效检测人源基因gapdh的mrna,1ng总rna中可以检出目的基因。最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,对于本领域的普通技术人员来说,在上述说明及思路的基础上还可以做出其它不同形式的变化或变动,这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。序列表<110>深圳市赛哲生物有限公司<120>一种基于解旋酶和切刻酶的等温扩增核酸检测方法及试剂盒<160>12<170>siposequencelisting1.0<210>1<211>174<212>dna<213>人(human)<400>1cgggtgatgcttttcctagattattctctggtaaatcaaagaagtgggtttatggaggtc60ctcttgtgtcccctccccgcagaggtgtggtggctgtggcatggtgccaagccgggagaa120gctgagtcatgggtagttggaaaaggacatttccaccgcaaaatggcccctctg174<210>2<211>22<212>dna<213>人(human)<400>2cgggtgatgcttttcctagatt22<210>3<211>19<212>dna<213>人(human)<400>3cagaggggccattttgcgg19<210>4<211>22<212>dna<213>人(human)<400>4ctgagtcatgggtagttggaaa22<210>5<211>186<212>dna<213>艰难梭菌(clostridiumdifficile)<400>5cacaagtggtagaagaaaggattgaagaagctaaaagcttaacttctgactctattaatt60atataaagaatgaatttaaactaatagaatctatttctgatgcactatacgatttaaaac120aacagaatgaattagaagagtctcattttatatcttttgaggatatatcggagactgatg180aaggct186<210>6<211>24<212>dna<213>艰难梭菌(clostridiumdifficile)<400>6cacaagtggtagaagaaaggattg24<210>7<211>21<212>dna<213>艰难梭菌(clostridiumdifficile)<400>7agccttcatcagtctccgata21<210>8<211>23<212>dna<213>艰难梭菌(clostridiumdifficile)<400>8aagagtctcattttatatctttt23<210>9<211>193<212>dna<213>人(human)<400>9acccattcattgaccttgattacatggtttacatgttccagtacgactccacccatggaa60agtacaagggtgaggttaaggcagaaggcggcaaactggtcattgatggtcatgcaatca120cagtctatagcgagagggacccagccaacattaagtggggtgatgcaggtgctacttatg180ttgtggagtctac193<210>10<211>20<212>dna<213>人(human)<400>10acccattcattgaccttgat20<210>11<211>20<212>dna<213>人(human)<400>11gtagactccacaacataagt20<210>12<211>22<212>dna<213>人(human)<400>12tggagtcgtactggaacatgta22当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1