光甘草定的制备方法及由该制备方法得到的光甘草定、化妆品与流程
本发明属于分离提纯
技术领域:
,具体涉及一种光甘草定的制备方法及由该制备方法得到的光甘草定、化妆品。
背景技术:
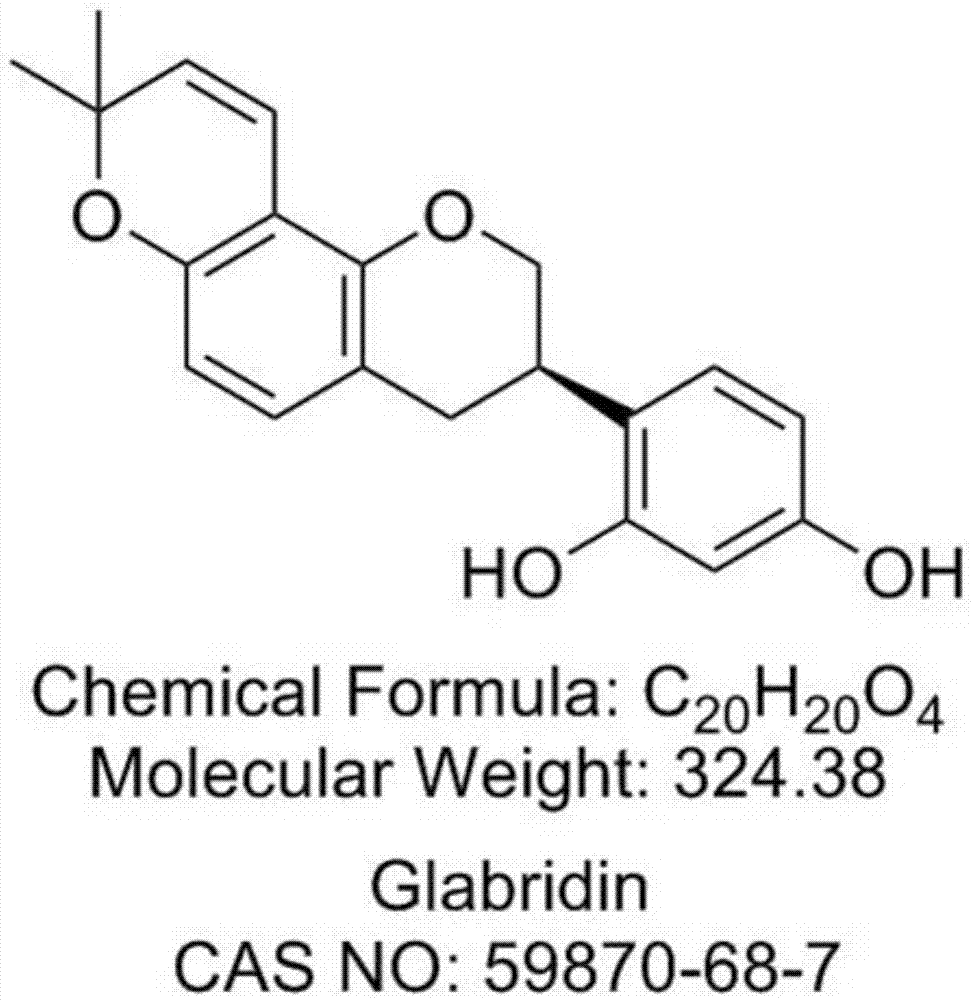
光甘草定是光果甘草(glycyrrhizaglabral.)中的主要黄酮类成分之一,现代药理研究表明,光甘草定具有调节免疫、降血脂、降血糖、类雌激素、抗菌、抗氧化、抗炎等作用。光甘草定在细胞色素p450/nadph氧化系统中显示出很强的抗自由基氧化作用,能明显抑制体内新陈代谢过程中所产生的自由基,以免受对氧化敏感的生物大分子(低密度脂蛋白ldl,dna)和细胞壁等被自由基氧化损伤,从而可以防治与自由基氧化有关的某些病理变化,如动脉粥样硬化,细胞衰老等。意大利研究还证实光甘草定有抑制食欲的作用,它能减少脂肪却又不减轻体重。光甘草定在心血管疾病的预防上具有良好的发展前景,在美容界,有着“美白黄金”的称号,足以说明光甘草定在医药、食品和美容领域的重要性。光甘草定的在光果甘草中的含量较少,仅有0.1%~0.3%,因此,需要一种从光果甘草中有效分离具有较高纯度的光甘草定的制备方法。专利cn102250107a、cn103360404a、cn105777771a、cn105294722a及cn104725394a公开了光甘草定的制备方法,但都使用了对环境不友好的有机溶剂,如乙酸乙酯等,易造成环境污染,不适合工业生产。专利cn107383039公开了的光甘草定的制备方法,虽然制备过程中不包括环境不友好的有机溶剂,但制备工艺繁琐,需要经过三个聚酰胺柱和一次硅胶柱,成本过高,不适合大规模生产。因此,亟需一种兼具工艺路线简单、环境友好、成本低、适合工业化生产的高纯度光甘草定的制备方法。鉴于此,特提出本发明。技术实现要素:本发明的目的在于提供一种光甘草定的制备方法;该制备方法工艺路线简单,制备过程中不使用环境不友好溶剂,环境友好,成本低,适合工业化生产。本发明的另一目的在于提供由上述制备方法制得的光甘草定;利用本发明方法制备得到的光甘草定具有较高的纯度。本发明的目的还在于提供一种化妆品,包括上述的光甘草定的制备方法制得的光甘草定。根据本发明的一个方面,本发明提供了一种光甘草定的制备方法,包括对水提得到的光果甘草渣进行醇提,得醇提取液,浓缩得醇提取物,然后依次利用大孔吸附树脂和聚酰胺柱层析分离得到含有光甘草定的洗脱液,重结晶得到光甘草定。作为进一步优选的技术方案,对粉碎的光果甘草进行水提,得到光果甘草渣;优选地,所述水提的料液比为1:6~12(w/w),优选为1:8~10(w/w);优选地,所述水提的温度为70~90℃,优选为80℃;优选地,所述水提的次数为2~4次,每次水提的时间为40~70min,优选为60min。作为进一步优选的技术方案,所述醇提所用的醇为c1-c4的醇,优选为甲醇或乙醇,进一步优选为乙醇;优选地,所述乙醇的体积分数为50%~95%,优选为80%~95%;优选地,所述醇提的料液比为1:5~20(w/v),优选为1:5~15(w/v),进一步优选为1:8~10(w/v);优选地,所述醇提温度为30~100℃,优选为50~100℃,进一步优选为60~80℃;优选地,所述醇提的次数为2~3次,每次0.5~6h,优选为1~4h,进一步优选为2h。作为进一步优选的技术方案,将醇提取液进行浓缩,得到提取物a,然后用醇溶解,分离得到液体部分,浓缩得到醇提取物;优选地,所述醇为c1-c4的醇,优选为乙醇,进一步优选为体积分数为50%~60%的乙醇;优选地,所述提取物a与醇的重量体积比为1:15~20g/ml。作为进一步优选的技术方案,所述依次利用大孔吸附树脂和聚酰胺柱层析分离得到含有光甘草定的洗脱液包括:(a)大孔吸附树脂柱层析分离:对浓缩得到的醇提取物进行洗脱,收集含有光甘草定的洗脱液b;(b)聚酰胺柱层析分离:对洗脱液b进行浓缩,得到提取物c,对提取物c进行洗脱,得到含有光甘草定的洗脱液。作为进一步优选的技术方案,步骤(a)中,所述大孔吸附树脂为非极性大孔吸附树脂、中极性大孔吸附树脂或极性大孔吸附树脂,优选为非极性大孔吸附树脂;优选地,步骤(a)中,所述大孔吸附树脂的孔径为80~200目;优选地,步骤(a)中,所述大孔吸附树脂的型号为ab-8、d101、ads-7或hp-300,优选为ab-8或d101;优选地,步骤(a)中,所述洗脱为梯度洗脱,梯度洗脱中所用洗脱剂依次为:35%~42%v/v乙醇、55%~62%v/v乙醇和68%~75%v/v乙醇,其中,醇提取物与洗脱剂的重量体积比依次为1:6~8g/ml、1:24~32g/ml和1:36~48g/ml;优选地,所述梯度洗脱中所用洗脱剂依次为40%v/v乙醇、60%v/v乙醇和70%v/v乙醇,其中,醇提取物与洗脱剂的重量体积比依次为1:6g/ml、1:32g/ml和1:48g/ml;优选地,步骤(a)中,所述洗脱剂的流速为250ml/min;优选地,步骤(a)中,所述收集含有光甘草定的洗脱液b是利用薄层色谱法收集rf为0.3~0.6的洗脱液,其中,所述薄层色谱法的展开剂为石油醚:乙酸乙酯=3:1。作为进一步优选的技术方案,步骤(b)中,所述聚酰胺的孔径为80~400目,优选为100~200目;优选地,步骤(b)中,所述洗脱为梯度洗脱,梯度洗脱中所用洗脱剂依次为:45%~52%v/v乙醇、55%~62%v/v乙醇和68%~75%v/v乙醇,其中,提取物c与洗脱剂的重量体积比依次为1:30~36g/ml、1:30~36g/ml和1:30~36g/ml;优选地,所述梯度洗脱中所用洗脱剂依次为50%v/v乙醇、60%v/v乙醇和70%v/v乙醇,其中,提取物c与洗脱剂的重量体积比依次为1:32g/ml、1:32g/ml和1:32g/ml;优选地,步骤(b)中,所述洗脱剂的流速为200ml/min;优选地,步骤(b)中,所述含有光甘草定的洗脱液是利用薄层色谱法收集rf为0.3~0.6的洗脱液,其中,所述薄层色谱法的展开剂为石油醚:乙酸乙酯=3:1。作为进一步优选的技术方案,对分离得到含有光甘草定的洗脱液进行浓缩,然后溶解在乙醇中,加入水,使得光甘草定重结晶,得到光甘草定;优选地,所述水与乙醇的体积比为3~7:3~7,优选为3~5:5~7。根据本发明的第二个方面,本发明提供了所述的光甘草定的制备方法制得的光甘草定。根据本发明的第三个方面,本发明提供了一种化妆品,包括所述的光甘草定的制备方法制得的光甘草定。本发明提供了一种光甘草定的制备方法,包括对水提得到的光果甘草渣进行醇提,得醇提取液,浓缩得醇提取物,然后依次利用大孔吸附树脂和聚酰胺柱层析分离得到含有光甘草定的洗脱液,重结晶得到光甘草定。该方法对经过水提的光果甘草渣进行醇提,可在醇提前去除光果甘草中大部分的水溶性杂质,经过两次特定的柱层析(大孔吸附树脂和聚酰胺柱层析)分离出纯度较高的光甘草定,再经过重结晶得到高纯度的光甘草定。本发明方法制备过程中没有使用环境不友好溶剂,环境友好,本发明经过醇提、两次柱层析和重结晶,即可制备得到高纯度的光甘草定,工艺路线简单,成本低,适合工业化生产。附图说明为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。图1为实施例1制备得到的光甘草定的结构图;图2为实施例1制备得到的光甘草定的esi-ms+图谱;图3为实施例1制备得到的光甘草定的1h-nmr图谱;图4为实施例1制备得到的光甘草定的13c-nmr图谱。具体实施方式下面将结合实施例及附图对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。根据本发明的一个方面,本发明提供了一种光甘草定的制备方法,包括对水提得到的光果甘草渣进行醇提,得醇提取液,浓缩得醇提取物,然后依次利用大孔吸附树脂和聚酰胺柱层析分离得到含有光甘草定的洗脱液,重结晶得到光甘草定。该方法对经过水提的光果甘草渣进行醇提,可在醇提前去除光果甘草中大部分的水溶性杂质,经过两次特定的柱层析(大孔吸附树脂和聚酰胺柱层析)分离出纯度较高的光甘草定,再经过重结晶得到高纯度的光甘草定。本发明方法制备过程中没有使用环境不友好溶剂,环境友好,本发明经过醇提、两次柱层析和重结晶,即可制备得到高纯度的光甘草定,工艺路线简单,成本低,适合工业化生产。本发明使用溶剂、填料都可循环使用,对环境影响极小,极大的节约了生产成本,是一种环境友好型生产工艺。作为进一步优选的技术方案,对粉碎的光果甘草进行水提,得到光果甘草渣;在该优选的实施方式中,通过水提除去光果甘草中的大部分水溶性杂质,有利于在简化柱层析分离过程的同时得到纯度更高的光甘草定;另外,对于水提得到的提取液,可分离得到水溶性的有效成分,这种方式在醇提前除去水溶性杂质的同时,利用得到的水提取物可用于分离出水溶性的有效成分,例如,甘草酸、甘草次酸和甘草苷等,进一步降低成本。作为进一步优选的技术方案,所述水提的料液比为1:6~12(w/w),典型但非限制性的料液比为1:6、1:7、1:8、1:9、1:10、1:11或1:12;在该优选的实施方式中,通过合理调整料液比,有效提取出水溶性成分,降低光果甘草渣中的水溶性杂质。作为进一步优选的技术方案,所述水提的料液比为1:8~10(w/w);在该优选的实施方式中,通过合理调整料液比,更有效地提取出水溶性成分。作为进一步优选的技术方案,所述水提的温度为70~90℃,典型但非限制性的温度为70℃、72℃、74℃、75℃、77℃、78℃、80℃、82℃、84℃、85℃、86℃、88℃、89℃或90℃;在该优选的实施方式中,通过合理调整水提温度,有效提取出水溶性成分,降低光果甘草渣中的水溶性杂质。作为进一步优选的技术方案,所述水提的温度为80℃。作为进一步优选的技术方案,所述水提的次数为2~4次,每次水提的时间为40~70min,优选为60min;典型但非限制性的次数为2次、3次或4次;典型但非限制性的每次水提的时间为40、42、44、46、47、49、50、52、53、55、57、59、60、62、63、65、67或70min;在该优选的实施方式中,通过合理调整水提的次数和时间,在兼顾成本的同时有效提取出水溶性成分,降低光果甘草渣中的水溶性杂质。作为进一步优选的技术方案,所述醇提所用的醇为c1-c4的醇,典型但非限制性的c1-c4的醇为甲醇、乙醇、乙二醇、1-丙醇、2-丙醇、1-丁醇、2-丁醇、1,1-丙二醇、1,2-丙二醇、2,2-丙二醇、1,3-丙二醇或1,1-丁二醇等;在该优选的实施方式中,利用c1-c4的低碳醇对光果甘草渣进行醇提,有效提取出光果甘草渣中的光甘草定,而且,低碳醇为环保型溶剂,环境友好。作为进一步优选的技术方案,所述醇提所用的醇为甲醇或乙醇。作为进一步优选的技术方案,所述醇提所用的醇为乙醇;在该优选的实施方式中,乙醇为低毒性,且更能有效提取出光果甘草渣中的光甘草定。作为进一步优选的技术方案,所述乙醇的体积分数为50%~95%,乙醇典型但非限制性的体积分数为50%、52%、54%、55%、58%、60%、63%、65%、67%、69%、70%、73%、75%、77%、79%、80%、82%、84%、86%、87%、89%、90%、92%、93%或95%等。作为进一步优选的技术方案,所述乙醇的体积分数为80%~95%;在该优选的实施方式中,体积分数为80%~95%的乙醇更能有效提取出光果甘草渣中的光甘草定。作为进一步优选的技术方案,所述醇提的料液比为1:5~20(w/v),典型但非限制性的料液比为1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:15、1:16、1:17、1:18、1:19或1:20;在该优选的实施方式中,通过合理调整料液比,有效提取出光果甘草渣中的光甘草定。作为进一步优选的技术方案,所述醇提的料液比为1:5~15(w/v),进一步优选为1:8~10(w/v);在该优选的实施方式中,通过合理调整料液比,在兼顾成本的同时,有效提取出光果甘草渣中的光甘草定。作为进一步优选的技术方案,所述醇提温度为30~100℃,优选为50~100℃,进一步优选为60~80℃;典型但非限制性的温度为30、32、34、35、37、38、40、42、44、45、47、48、50、52℃、54、55、57、58、60、62、64、65、67、68、70、72、74、75、77、78、80、82、84、85、86、88、89、90、92、94、95、96、98、99或100℃。作为进一步优选的技术方案,所述醇提的次数为2~3次,每次0.5~6h;典型但非限制性的每次水提的时间为0.5、1、1.5、2、2.5、3、3.5、4、4.5、5、5.5或6h。作为进一步优选的技术方案,每次水提的时间为1~4h;在该优选的实施方式中,通过合理调整醇提的时间,在降低成本的同时有效提取出光果甘草渣中的光甘草定。作为进一步优选的技术方案,每次水提的时间为2h;在该优选的实施方式中,通过合理调整醇提的时间,更好地兼具成本和提取效率。作为进一步优选的技术方案,将醇提取液进行浓缩,得到提取物a,然后用醇溶解,分离得到液体部分,浓缩得到醇提取物。在本发明优选的实施方式中,通过对醇提取液浓缩得到的提取物a溶解后,再分离,除去一部分杂质,得到的液体部分中光甘草定的纯度提高。需要说明的是,对于浓缩没有特殊的限制,采用本领域常用的减压浓缩除去醇溶剂得到提取物即可,减压得到的醇溶剂可回收使用,进一步降低了成本。对于溶解没有特殊的限制,采用本领域常用的超声溶解使得提取物a能够溶解即可;对于分离没有特殊的限制,采用本领域常用的过滤除去不溶性杂质即可。作为进一步优选的技术方案,所述醇为c1-c4的醇,典型但非限制性的c1-c4的醇为甲醇、乙醇、乙二醇、1-丙醇、2-丙醇、1-丁醇、2-丁醇、1,1-丙二醇、1,2-丙二醇、2,2-丙二醇、1,3-丙二醇或1,1-丁二醇等。在该优选的实施方式中,c1-c4的低碳醇可使得提取物a中的光甘草定有效溶解。作为进一步优选的技术方案,所述醇为乙醇,进一步优选为体积分数为50%~60%的乙醇;乙醇典型但非限制性的体积分数为50%、52%、54%、55%、58%或60%。作为进一步优选的技术方案,所述提取物a与乙醇的重量体积比为1:15~20g/ml;典型但非限制性的重量体积比为1:15g/ml、1:15.5g/ml、1:16g/ml、1:16.5g/ml、1:17g/ml、1:18g/ml、1:19g/ml或1:20g/ml。在该优选的实施方式中,通过合理调整提取物a与乙醇的重量体积比,有效溶解提取物a。作为进一步优选的技术方案,所述依次利用大孔吸附树脂和聚酰胺柱层析分离得到含有光甘草定的洗脱液包括:(a)大孔吸附树脂柱层析分离:对浓缩得到的醇提取物进行洗脱,收集含有光甘草定的洗脱液b;(b)聚酰胺柱层析分离:对洗脱液b进行浓缩,得到提取物c,对提取物c进行洗脱,得到含有光甘草定的洗脱液。该方法只需两次柱层析,依次为大孔吸附树脂和聚酰胺柱层析,首先利用大孔树脂吸附容量较大的优势去除大部分杂质,得到较高纯度的含有光甘草定的洗脱液,另外,大孔树脂吸附选择性良好,吸附快解析也快,分离效果明显;再利用聚酰胺柱层析与光甘草定的酚羟基形成氢键缔合产生吸附,得到纯度更高的含有光甘草定的洗脱液。需要说明的是,在步骤(a)和步骤(b)中,洗脱前需要进行拌样和装柱,采用本领域技术人员常用的拌样方式进行拌样和装柱即可。例如,步骤(a)中,拌样时,醇提取物与大孔吸附树脂的重量比为1:45~55;步骤(b)中,拌样时,提取物c与聚酰胺的重量比为1:6~8。作为进一步优选的技术方案,步骤(a)中,所述大孔吸附树脂为非极性大孔吸附树脂、中极性大孔吸附树脂或极性大孔吸附树脂,作为进一步优选的技术方案,所述大孔吸附树脂为非极性大孔吸附树脂;在该优选的实施方式中,采用非极性大孔吸附树脂更有利于分离出光甘草定。作为进一步优选的技术方案,步骤(a)中,所述大孔吸附树脂的孔径为80~200目;典型但非限制性的孔径为80、90、100、110、120、130、140、150、160、170、180、190或200目。作为进一步优选的技术方案,步骤(a)中,所述大孔吸附树脂的型号为ab-8、d101、ads-7或hp-300。作为进一步优选的技术方案,所述大孔吸附树脂的型号为ab-8或d101;在该优选的实施方式中,ab-8型大孔吸附树脂是苯乙烯型弱极性共聚体,d101型大孔吸附树脂是苯乙烯型非极性共聚体,能够更有效地分离出光甘草定。作为进一步优选的技术方案,步骤(a)中,所述洗脱为梯度洗脱,梯度洗脱中洗脱剂和洗脱剂用量为:洗脱剂醇提取物与洗脱剂的重量体积比体积分数35%~42%乙醇1:6~8体积分数55%~62%乙醇1:24~32体积分数68%~75%乙醇1:36~48在本发明优选的实施方式中,通过梯度洗脱以及合理调整梯度洗脱的洗脱剂和洗脱剂的用量,更有效地分离出光甘草定。作为进一步优选的技术方案,步骤(a)中,梯度洗脱中洗脱剂和洗脱剂用量为:洗脱剂醇提取物与洗脱剂的重量体积比体积分数40%乙醇1:6体积分数60%乙醇1:32体积分数70%乙醇1:48在本发明优选的实施方式中,通过合理调整梯度洗脱的洗脱剂和洗脱剂的用量,更有效地分离出光甘草定,有利于得到纯度更高的光甘草定。作为进一步优选的技术方案,步骤(a)中,所述洗脱剂的流速为250ml/min。作为进一步优选的技术方案,步骤(a)中,所述收集含有光甘草定的洗脱液b是利用薄层色谱法收集rf为0.3~0.6的洗脱液,其中,所述薄层色谱法的展开剂为石油醚:乙酸乙酯=3:1。在该优选的实施方式中,利用薄层色谱法收集光甘草定含量较高的洗脱液。rf是比移值,是薄层色谱法中原点到斑点中心的距离与原点到溶剂前沿的距离的比值。是色谱法中表示组分移动位置的一种方法的参数。作为进一步优选的技术方案,步骤(b)中,所述聚酰胺的孔径为80~400目,优选为100~200目;典型但非限制性的孔径为80、90、100、110、120、130、140、150、160、170、180、190、200、220、240、250、270、290、300、320、340、350、370、390或400目。作为进一步优选的技术方案,步骤(b)中,所述洗脱为梯度洗脱,梯度洗脱中洗脱剂和洗脱剂用量为:洗脱剂提取物c与洗脱剂的重量体积比体积分数45%~52%乙醇1:30~36体积分数55%~62%乙醇1:30~36体积分数68%~75%乙醇1:30~36在本发明优选的实施方式中,通过梯度洗脱以及合理调整聚酰胺柱层析梯度洗脱的洗脱剂和洗脱剂的用量,更有效地分离出光甘草定。作为进一步优选的技术方案,步骤(b)中,梯度洗脱中洗脱剂和洗脱剂用量为:洗脱剂提取物c与洗脱剂的重量体积比体积分数50%乙醇1:32体积分数60%乙醇1:32体积分数70%乙醇1:32在本发明优选的实施方式中,通过合理调整聚酰胺柱层析梯度洗脱的洗脱剂和洗脱剂的用量,更有效地分离出光甘草定,有利于得到纯度更高的光甘草定。作为进一步优选的技术方案,步骤(b)中,所述洗脱剂的流速为200ml/min;作为进一步优选的技术方案,步骤(b)中,所述含有光甘草定的洗脱液是利用薄层色谱法收集rf为0.3~0.6的洗脱液,其中,所述薄层色谱法的展开剂为石油醚:乙酸乙酯=3:1。在该优选的实施方式中,利用薄层色谱法收集富含光甘草定的洗脱液。作为进一步优选的技术方案,对分离得到含有光甘草定的洗脱液进行浓缩,然后溶解在乙醇中,加入水,使得光甘草定重结晶,得到光甘草定;在该优选的实施方式中,通过将浓缩后的洗脱液溶解在乙醇中,之后加入水,使得光甘草定重结晶,得到纯度更高的光甘草定,重结晶方法可使得光甘草定快速结晶的同时具有较高的纯度。需要说明的是,上述溶解是指达到饱和的状态,对于溶解的温度没有特殊限制,20~30℃的室温即可。作为进一步优选的技术方案,所述水与乙醇的体积比为3~7:3~7,优选为3~5:5~7。按体积计,水典型但非限制性的体积份为3份、4份、5份、6份或7份,乙醇典型但非限制性的体积份为3份、4份、5份、6份或7份。需要说明的是,制备方法的多次浓缩步骤中,得到的固体部分含有目标产品,除去的醇可回收使用,制备过程中对环境的影响极小,极大的节约了生产成本,是一种环境友好型生产工艺。根据本发明的第二个方面,本发明提供了所述的光甘草定的制备方法制得的光甘草定。本发明制备得到的光甘草定纯度较高。根据本发明的第三个方面,本发明提供了一种化妆品,包括所述的光甘草定的制备方法制得的光甘草定;本发明制备得到的光甘草定纯度高,具有良好的抗炎美白的功效,具有抑制黑色素的作用,可应用于化妆品。需要说明的是,本发明中“v/v”是指具有相同单位的体积比,同样的,“w/w”是指具有相同单位的重量比;本发明中“w/v”是指g/ml,也可以理解为与g/ml同等意义的kg/l。下面将结合实施例和对比例对本发明的技术方案进行进一步地说明。实施例1光甘草定的制备一种光甘草定的制备方法,包括以下步骤:(1)取粉碎的光果甘草1kg,依次利用10重量份水、8重量份水和8重量份水在80℃下提取三次,每次60min;得到的提取液另做处理,收取得到的光果甘草渣。(2)取步骤(1)得到的光果甘草渣,依次用10重量份95%v/v乙醇、8重量份95%v/v乙醇和8重量份95%v/v乙醇在70℃下提取三次,每次2h,过滤,合并得醇提取液。(3)取步骤(2)得到的醇提取液,减压浓缩回收乙醇,得到110g提取物a,加入55%v/v乙醇2l,进行超声溶解,过滤得到液体部分,减压浓缩得到醇提取物。(4)大孔吸附树脂柱层析分离:将步骤(3)得到的醇提取物与6kg的ab-8(140目)大孔吸附树脂拌样,装柱,以250ml/min的速度进行洗脱(洗脱条件如表1所示),利用薄层色谱法(展开剂为石油醚:乙酸乙酯=3:1)收集rf为0.3~0.6的洗脱液,收集含有光甘草定的洗脱液b。表1大孔吸附树脂的洗脱条件洗脱剂醇提取物与洗脱剂的重量体积比体积分数40%乙醇1:6g/ml体积分数60%乙醇1:32g/ml体积分数70%乙醇1:48g/ml(5)聚酰胺柱层析分离:对洗脱液b进行减压浓缩,得到提取物c,与600g聚酰胺拌样,装柱,以200ml/min的速度进行洗脱(洗脱条件如表2所示),利用薄层色谱法(展开剂为石油醚:乙酸乙酯=3:1)收集rf为0.3~0.6的洗脱液,得到含有光甘草定的洗脱液。表2聚酰胺柱层析的洗脱条件洗脱剂提取物c与洗脱剂的重量体积比体积分数50%乙醇1:32g/ml体积分数60%乙醇1:32g/ml体积分数70%乙醇1:32g/ml(6)对分离得到含有光甘草定的洗脱液进行减压浓缩,然后将浓缩得到的固体部分溶解在乙醇中,滴加水(加水量与乙醇的体积比为4:6),使得光甘草定重结晶,得到1.1g纯度为98.5%的光甘草定。实施例2将实施例1中光果甘草的用量增大50倍,增加到50kg,其余原料的用量以相应的倍率增大,最终得到14g纯度为97.8%的光甘草定。本发明的制备方法进行大规模生产后,制备得到的光甘草定依然具有很高的纯度。实施例3~15实施例3~15与实施例1的不同之处在于步骤(2)醇提的具体工艺,具体不同之处以及最终制备得到的光甘草定的重量和纯度如表3所示。表3实施例3~15不同于实施例1的醇提的具体工艺乙醇v/v温度℃次数每次时间h重量g纯度%实施例330%———1.2872.3实施例450%———1.1185.1实施例580%———1.1696.9实施例6—30——0.9270.6实施例7—100——1.1497.9实施例8—50——1.1382.9实施例9—60——1.1194.3实施例10—80——1.1298.1实施例11——2—0.7398.9实施例12———0.50.6298.7实施例13———61.6192.9实施例14———10.8698.1实施例15———41.4593.3表3中,“—”代表与实施例1相同。实施例16~18实施例16~18与实施例1的不同之处在于步骤(2)醇提的料液比,具体不同之处以及最终制备得到的光甘草定的重量和纯度如表4所示。表4实施例16~18的料液比及对应的光甘草定的重量和纯度实施例19~20实施例19~20与实施例1的不同之处在于步骤(4)大孔吸附树脂的梯度洗脱所用乙醇洗脱剂的体积分数,具体不同之处以及最终制备得到的光甘草定的重量和纯度如表5所示。表5实施例19~20大孔吸附树脂的乙醇洗脱剂的体积分数乙醇乙醇乙醇重量g纯度%实施例1935%55%75%1.0587.7实施例2042%62%68%1.0688.4实施例21~22实施例21~22与实施例1的不同之处在于步骤(4)聚酰胺柱层析的梯度洗脱所用乙醇洗脱剂的体积分数,具体不同之处以及最终制备得到的光甘草定的重量和纯度如表6所示。表6实施例21~22聚酰胺柱层析的乙醇洗脱剂的体积分数乙醇乙醇乙醇重量g纯度%实施例2445%55%75%0.8185.91实施例2552%62%68%0.8286.12对比例1对比例1与实施例1的不同之处在于,无水提,即无实施例1中的步骤(1),即将实施例1步骤(2)中的光果甘草渣直接替换为1kg粉碎的光果甘草,然后直接用于醇提,得到0.82g纯度为56.10%的光甘草定。对比例2对比例2与实施例1的不同之处在于,无大孔吸附树脂柱层析分离,即无实施例1中的步骤(4),即将实施例1步骤(5)中的提取物c直接替换为醇提取物,然后直接进行聚酰胺柱层析分离,得到0.54g纯度为31.61%的光甘草定。对比例3对比例3与实施例1的不同之处在于,无聚酰胺柱层析分离,即无实施例1中的步骤(5),即将实施例1步骤(4)中的醇提取物直接溶解在乙醇中进行重结晶,得到0.9g纯度为27.09%的光甘草定。对比例4对比例4与实施例1的不同之处在于,先进行聚酰胺柱层析分离,再进行大孔吸附树脂柱层析分离,得到0.31g纯度为42.10%的光甘草定。试验例1对实施例1制备得到的光甘草定(结构如图1所示)进行esi-ms+和1h-nmr、13c-nmr检测,得到的结果如图2、图3和图4所示。图2是实施例1制备得到的光甘草定的esi-ms+图谱。由图谱中m/z360[m+2nh4]+可知,供试品分子量为324,与图1所示结构一致。图3是实施例1制备得到的光甘草定的1h-nmr图谱。图谱中,δ1.42和1.44[6h,s,s,(ch3)2]、2.77(1h,dd,j=15.6,6.5hz,h-4α)、2.91(1h,dd,j=15.6,11.2hz,h-4β)、3.45(1h,m,h-3)、3.93(1h,t,j=10.3hz,h-2β)、4.31(1h,m,h-2α)、5.49(1h,d,j=9.9hz)、6.25~6.29(3h,m)、6.58(1h,d,j=9.9hz)、6.75(1h,d,j=8.2hz)、6.83(1h,d,j=8.2hz),均为光甘草定的特征谱峰,与图1所示结构一致。图4是实施例1制备得到的光甘草定的13c-nmr图谱。图谱中,δ27.2(ch3)、27.4(ch3)、30.4(ch2,c-4)、31.4(ch,c-3)、70.1(ch2,c-2)、75.5(c,c-2”)、102.4(ch,c-6)、106.6(ch,c-3')、108.3(ch,c-5')、109.7(c,c-8)、114.8(c,c-10)、116.9(ch,c-4”)、118.9(c,c-1')、127.7(ch,c-3”)、128.7(ch,c-6')、129.1(ch,c-5)、149.7(c,c-2')、151.4(c,c-7)、155.6(c,c-4')、155.9(c,c-9),均为光甘草定的特征谱峰,与图1所示结构一致。从图2、图3和图4所示的esi-ms+和1h-nmr、13c-nmr数据可知,实施例1得到的化合物即为光甘草定。试验结果表明,本发明制备方法工艺路线简单,制备过程中不使用环境不友好溶剂,环境友好,成本低,适合工业化生产,制备得到的光甘草定纯度较高。最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1