利用CRISPR-Cas9构建的斑马鱼msi1基因突变体及其构建方法与流程
本发明涉及分子生物学领域,特别涉及一种利用crispr-cas9构建斑马鱼msi1基因突变体的方法以及使用该方法构建的斑马鱼msi1基因突变体。
背景技术
musashirna结合蛋白1型(musashirnabindingprotein1)是一种由msi1基因编码的蛋白。该蛋白包含两个保守的串联rna识别基序,并作为rna结合蛋白参与基因的转录后调控。通过调节转录后的翻译过程来使干细胞保持未分化状态,通过调节notch,wnt/β-catenin等信号通路影响干细胞的增殖和细胞命运的决定以及肿瘤的形成,是一种控制自我更新和终末分化之间平衡的干细胞标记物。msi1在神经祖细胞中高度表达,其对于大脑的正常发育十分重要。该基因的突变导致常染色体隐性原发性小头畸形。此外,该基因的过表达与胶质瘤和黑素瘤的恶性程度和增殖活性也有关系。而降低msi1基因的表达水平可有效诱导癌症细胞凋亡,阻止有丝分裂和细胞周期进程,抑制肿瘤细胞增殖并导致肿瘤消退。研究msi1的功能对临床肿瘤疾病的深入探究和诊断治疗可能提供新的途径。
斑马鱼是一种用于研究脊椎动物胚胎学和发育遗传学的模式生物,基于其快速迭代的优势,可以很好地用于癌症肿瘤学的研究。目前在斑马鱼中对于msi1基因功能的研究仍是有限的,本专利通过crspr-cas9建立了斑马鱼msi1的突变体,为进一步研究msi1的功能提供了材料基础。
crispr/cas9是细菌和古细菌在长期演化过程中形成的一种适应性免疫防御,可用来对抗入侵的病毒及外源dna。crispr是指规律成簇的间隔短回文重复序列(clusteredregularlyinterspacedshortpalindromicrepeat,crispr),最早是由日本科学家ishino在大肠杆菌中发现。cas则指的是crispr相关蛋白,一般定位于crispr位点附近,是一个有多种亚型多肽的蛋白家族,而cas9是应用最为广泛的一个亚型。2013年1月,woongyhwang等首次将该技术应用于斑马鱼基因编辑中。通过外源导入靶向基因组的grna和cas9mrna,grna即可引导cas9蛋白切割基因组靶点附近的dna双链,进而引发细胞的错配修复,从而造成基因突变。
技术实现要素:
本发明目的在于构建了斑马鱼msi1基因的突变体。
为实现上述构建斑马鱼msi1基因突变体的目的,本发明提供的技术方案是:
1.根据基因序列设计cas9靶位点;
2.确认靶位点在斑马鱼基因组中的特异性;
3.显微共注射msi1-grna和cas9mrna到斑马鱼胚胎,并验证突变效率;
4.饲养f0胚胎至性成熟,和野生型外交,检测f1胚胎突变类型;
5.饲养f1胚胎至1月龄,鉴定突变类型,相同突变类型集中饲养;
6.相同突变类型f1成鱼自交,得到f2胚胎,生长到1月龄鉴定纯合子;
7.得到2种突变类型的纯合f2成鱼,自交获得稳定遗传的突变体后代。
借助本发明,至少可以达到以下有益效果:
1.获得msi1基因纯合突变体;
2.为研究msi1基因功能提供材料;
3.该基因突变体可用于肿瘤细胞抑制研究;
4.该基因突变体对临床肿瘤疾病的深入探究和诊断治疗可能提供新的途径。
上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合符图详细说明如下。
附图说明
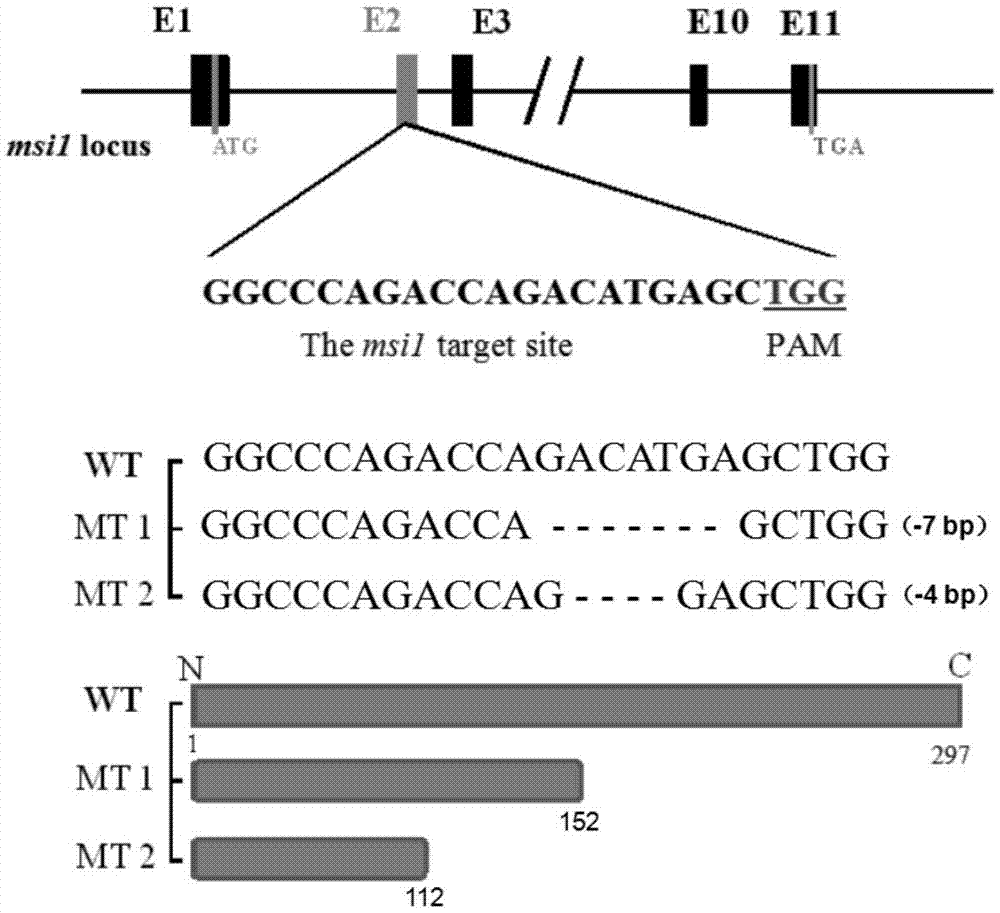
图1为msi1突变体构建示意图。图示msi1突变体构建示意图,显示了msi1基因座21个外显子,设计cas9靶位点位于第2个外显子上,绿色竖线指示翻译起始密码子atg,蓝色竖线指示终止密码子tga;中间显示了靶位点突变类型,分别为缺失几个碱基和缺失碱基;下图显示预测的氨基酸长度,野生型蛋白氨基酸为297个,突变体的msi1蛋白翻译都提前终止,缺失7个碱基和4个碱基突变类型对应氨基酸分别为152个和112个。
图2为msi1-grna的电泳条带结果图。图示msi1-grna的电泳条带结果,条带单一,且符合预期大小,约130bp。
图3为斑马鱼msi1基因第二个外显子序列。本发明构建突变体所设计的cas9靶位点位于此外显子上。
图4为msi1基因cas9靶序列。图示本本发明构建突变体所设计的cas9靶位点序列。
图5为msi1突变位点pcr鉴定序列。图示本发明构建突变体所设计的pcr鉴定序列,序列长度为130bp。
图6为msi1基因鉴定引物p1序列。图示本发明构建突变体所设计的pcr鉴定上游引物。
图7为msi1基因鉴定引物p2序列。图示本发明构建突变体所设计的pcr鉴定下游引物。
图8为msi1基因grna合成引物p3序列。图示本发明构建突变体所设计grna合成上游引物。
图9为msi1基因序列。图示本发明构建突变体的基因序列,其中蓝色序列为蛋白编码序列,位于外显子上;灰色序列为内含子序列;褐色序列为外显子非翻译区。
具体实施方式
下面结合附图和实施例,对本发明具体实施方式作进一步详细的描述。以下实施例用于说明本发明,但不用于限制本发明的范围。
实施例1
本发明一较佳实施例提供一种利用crispr-cas9构建斑马鱼msi1基因突变体的方法,具体包括以下步骤:
1.首先,设计msi1基因cas9靶位点,方法如下:
a.从ensemble网站上获取msi1的蛋白编码序列(cds),如附图9所示;
b.运用http://zifit.partners.org/zifit/csquare9nuclease.aspx网站设计靶位点,打开网站,输入msi1蛋白编码序列;
c.设定靶位点长度为20个碱基,靶点3’端的3个碱基构成pam区,序列为ngg(n为任意碱基),输出靶位点序列;
d.msi1基因的cas9靶位点序列为:
5’-ggcccagaccagacatgagctgg-3’,为正义链靶点,位于第2个外显子,如附图3-4所示。
2.进一步的,所述的确认msi1-cas9靶位点的方法如下:
a.在ensemble网站上比对靶位点序列,比对结果显示靶点是msi1基因的单一位点;
b.在靶位点上下游设计引物,
5’端引物为5’-taattttgatctctctgttgtc-3’,如附图6所示,
3’端引物为,5’-ttaatatgattaaaactcac-3’,如附图7所示,产物大小为123个碱基对,从野生型斑马鱼基因组中pcr扩增这段序列,其中应包含靶点序列,pcr程序为:95℃预变性5min,95℃变性30sec,60℃退火30sec,72℃延伸10sec,40循环。pcr扩增产物2%琼脂糖凝胶电泳,大小符合预期;
c.将pcr产物进行测序,测序结果和预测靶点序列一致。
3.进一步的,制备msi1-grna,具体步骤如下:
a.根据靶位点设计grna体外转录模板的上游引物,命名为p3,序列为,5’-gatcactaatacgactcactataggcccagaccagacatgagcgttttagagctagaaat-3’,如附图8所示,下游引物使用pt7-grna质粒上的通用引物,命名为p4。
b.用p3和p4引物对,以体外转录载体pt7-grna质粒为模板,使用高保真酶pcr扩增得到msi1-grna体外转录模板,pcr体系为:模板1微升,高保真缓冲液25微升,dntps12微升,高保真酶1微升,引物各1微升,双蒸水补齐至50微升。pcr条件为:98℃预变性3min,98℃变性30sec,60℃退火30sec,68℃延伸30sec,40循环。取1微升pcr产物电泳,结果确认为单一条带(130bp),回收pcr产物,用酚-氯仿法纯化后得到转录模板。
c.用t7转录试剂盒体外转录msi1-grna,反应体系为:模板1微克,ntp4微升,缓冲液2微升,t7酶1微升,depc水补至20微升。反应条件为37℃,1h;然后加入1微升dna酶(turbodnase),37℃,15min以去除dna模板。取1微升转录产物电泳,产物大小约100bp左右,如附图2所示,符合预期,用酚-氯仿法纯化回收得到grna。
d.酚-氯仿法纯化方法为:将回收产物用depc水补齐至200微升,按体积比1:1加入酚-氯仿,振荡混匀后12,000g离心10min,取上清液于新的去rna酶1.5毫升离心管中,再加入2.5倍体积的异丙醇,颠倒混匀后置于-80℃,静置30min以上后取出,12,000g离心15min,去掉上清液,留下白色沉淀即为grna,加入200微升75%的乙醇清洗grna,随后除去乙醇,将离心管开盖置于超净工作台数分钟,晾干grna后加入30微升depc水溶解,将得到的grna溶液保存置-80℃备用。
4.进一步的,制备cas9mrna,具体步骤如下:
a.用限制性内切酶线性化psp6-spcas9载体,取0.5微升酶切产物电泳,确认线性化完全后回收,并进行纯化处理。
b.用sp6体外转录试剂盒转录线性化载体,转录体系为:10微升2xntp/cap,2微升10x缓冲液,2微升sp6转录酶,1微克载体,depc水补齐至20微升。反应条件为,37℃,2h,随即加入1微升dna酶,去除dna模板。纯化回收得到cas9mrna,取0.5微升产物电泳,1%琼脂糖凝胶,产物大小约2,000bp。
c.cas9mrna添加ploya序列,体系为:回收的mrna,5微升10mmatp,5微升10x缓冲液,1微升e.colipoly(a)聚合酶,depc水补至50微升。反应条件为,37℃,1h。纯化回收得到cas9mrna-ploya,取0.5微升产物电泳,1%琼脂糖凝胶,产物大小约2,000bp。
5.进一步的,所述的制备rna实验中,各部分实验所用耗材和试剂都是去rna酶的,实验中未明确表明温度的,产物和试剂应保持在4℃以下,避免rna受热降解。
6.进一步的,所述的制备f0斑马鱼并检测msi1基因靶点突变效率,通过显微注射和pcr测序来完成,其具体方法如下:
a.预先配置注射体系,包括cas9mrna和msi1-grna,加入1/10体积酚红,作为指示剂,注射到单细胞期胚胎中,注射约200枚胚胎,注射剂量为cas9mrna300pg,grna50-100pg。
b.取注射后第2天发育正常的胚胎10枚,碱裂解法提取基因组dna:将胚胎置于pcr管中,加入100微升50mmnaoh溶液,pcr仪加热到95℃,30min裂解胚胎,取出后振荡均匀,加入1/10体积ph8.0的tris-hcl中和naoh。
c.用引物p1和p2扩增基因组dna,将pcr产物通过ta克隆连接至t载体上,测序检测突变类型,结果显示有两种突变类型,分别为缺失7个碱基和4个碱基,如附图1所示。将突变效率较高的f0胚胎饲养起来,用于后续检测筛选可遗传的突变。
7.进一步的,所述的检测f0斑马鱼生殖遗传的方法如下:
a.取性成熟的f0成鱼和野生型成鱼外交,得到f1胚胎,随机挑选5-10个胚胎混合提取基因组dna。
b.用pcr扩增靶位点片段,pcr产物通过ta克隆测序,确定其突变类型。表明f0斑马鱼msi1基因突变可以遗传,随后饲养可遗传突变的f1,以备后续筛选。
8.进一步的,所述的确认f1斑马鱼msi1基因突变类型,方法如下:
a.f1长到1-2月龄,剪取尾鳍,提取基因组dna。
b.通过pcr分析和ta克隆测序,进一步确认f1斑马鱼msi1基因突变类型,筛选得到2种突变类型的f1斑马鱼,命名为mt-1和mt-2,相同突变类型的f1可以混合饲养。
9.进一步的,所述的筛选f2斑马鱼msi1基因纯合突变体,具体方法是:将相同突变的f1成鱼进行自交,得到f2胚胎,饲养至1-2月龄,通过剪尾鳍,提基因组,pcr,测序确认f2斑马鱼纯合突变。得到相同突变的纯合子后,通过自交即可获得大量纯合突变后代,即建立起了可稳定遗传的msi1基因突变体。
实施例2
本发明一较佳实施例提供一种利用crispr-cas9构建的斑马鱼msi1基因突变体,其具体特征在于:
1.所述的msi1基因突变体包含两个品系,具有不同的突变类型;
2.所述的msi1基因突变体,其突变类型分别为缺失7个碱基和4个碱基;
3.所述的msi1基因突变体,突变缺失碱基位于靶位点附近;
4.所述的msi1基因突变体,其突变序列如附图1所示;
5.所述的msi1基因突变体,其蛋白翻译提前终止,野生型msi1蛋白有896个氨基酸,而突变体中,缺失7个碱基和4个碱基分别对应了426个氨基酸和258个氨基酸。
6.所述的msi1基因突变体,为进一步研究msi1的功能提供了材料基础,有助于在癌症治疗的预后和功能相关性等方面发挥作用。
以上所述仅是本发明的优选实施方法,并不用于限制本发明,应当指出,对于本发明领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变形,这些改进和变形也应视为本发明的保护范围。
本发明由国家自然科学基金(青年科学基金项目——81502500)、江苏省自然科学基金(青年科学基金项目——bk20150293)及苏州市民生科技(医疗卫生应用基础研究项目——sys2018074)提供资助。
- 还没有人留言评论。精彩留言会获得点赞!