一种基因组组装方法与流程
本发明涉及一种基因组组装方法,属于基因组组装技术领域。
背景技术:
测序仪通过对基因组片段的测序产生了随机的读出序列片段(读段)。这些读段在基因组上的分布是随机的。基因组组装的过程就是把这些读段按照正确的顺序排列和连接,组装成碱基连续的dna序列片段(连续片段),最终复原整条染色体及整个基因组的序列。这个组装的过程一般包括三步:连续片段的组装,有缺口的非连续片段的组装,缺口的补齐。基因组组装的困难来源于基因组存在的大量重复序列(即长度不定、序列相似或一样的两个/段或多个/段序列)。此外测序仪在实际测出读段的过程中会产生错误,导致原来不一样的序列变得一样。若是没有重复序列和测序错误,基因组的组装就是一个很简单的任务。重复序列在基因组中可分为两个大类:串联重复序列和散布重复序列。串联重复是一组头尾直接相连的非常相似的重复单位组成的序列,通过局部重复产生。典型的串联重复序列包括rdna、着丝粒重复序列等。散布重复序列是分布于基因组中不同位置的非局部重复序列。在有些重复序列中,串联重复和非串联重复序列都有,这些区域很长,形成复杂重复序列。测序产生的来源于不同重复序列拷贝的读段具有序列上的相似性。目前单分子测序读段的长度n50一般大于10-15kb,最长达到了100kb以上。若是一个重复序列加上其两端的单拷贝序列一起被一条读段全部覆盖,则这个区域不存在组装的问题。当前需要解决的重点是超出了读段平均或n50长度的重复序列的组装问题。但是由于重复序列的存在,而且由于测序错误,导致单拷贝序列的组装在现有方法中也面临组装效率不高的问题。
对于长单分子测序数据现在最常用的基因组组装方法采用了基于overlap-layout-consensus(olc)(myersetal,2000,science,287,2196–2204.)或字符串图(stringgraph,sg)(myers2005,bioinformatics,21,suppl2,ii79-ii85)的策略。olc方法也可以简洁地用sg来描述,我们统称为sg类方法。现有的sg类方法常用软件包括pbcr(berlinetal.2015,nat.biotechnol.33,623–30)、canu(korenetal.2017,genomeres.27,722–736)、falcon(chinetal.2016,nat.methods13,1050–1054)、mecat(xiaoetal.2017,nat.methods.doi:10.1038/nmeth.4432)等。sg方法中的关键是利用可传递性简化路径(transitivereduction,tu)的方法来去除多余的读段(所有的序列特别相似的读段被压缩成一条)。即在构建所有读段的重叠图后,利用tu将很多节点的进出边数各简化到一条。这样在很多路径上就会没有分支。若是一个读段节点在简化后图中的进或出重叠边度数大于1,则称之为交叉节点,其他的节点为内部节点。没有交叉节点的一个通路就可以形成一条连续片段,在sg中可被进一步压缩到一起。交叉节点代表了单拷贝序列区域和重复序列区域的连接之处(这个节点上的读段包括了两种类型序列的各一部分;测序仪在测出读段的时候会犯错误,导致其测出带有测序错误的读段,这些序列错误包括碱基的插入、缺失、变异,或是来源于不同位置的序列的嵌合体,这些错误也可能导致交叉节点序列。由于测序错误的存在,导致难以形成一个统一的标准来区分读段之间的差异到底是由测序错误引起,还是来源于重复序列的不同拷贝而引起的)。在这个路径简化的过程中,单拷贝区域被简化成一长串读段形成的单一路径,连接到一起形成单拷贝序列连续片段;而一段重复序列也可以被压缩成一串读段形成的单一路径,形成重复序列连续片段。由于来源于不同重复序列拷贝的读段会被压缩到一起,导致不同拷贝的重复序列变成一个,因而不能区分开。但是由于交叉节点的存在,形成的重复序列连续片段在被压缩的起点和终点位置断开,导致组装出的序列片段的碎片化,进而导致无法真正复原整个原始基因组序列。而且现有方法对读段组装过程的处理步骤过多,导致处理速度慢、软件的复杂程度高。
技术实现要素:
本发明的目的在于,提供一种快速高效的基因组组装方法,它可以有效解决现有技术中存在的问题,尤其是现有技术中通过对读段的压缩来寻找组装图中没有交叉节点的路径而形成序列连续片段,但在此过程中由于操作步骤过多,导致组装速度慢、软件复杂度高;也由于在将相似的多段重复序列压缩过程中,很多来源于不同重复序列拷贝的读段会被压缩到一起,导致不同拷贝的重复序列变成一个,因而不能区分开;而且由于相似序列或测序错误导致交叉节点的存在,形成的序列片段在被压缩的起点和终点位置断开,导致组装出的序列出现连续片段的碎片化的问题。
为解决上述技术问题,本发明采用如下的技术方案:
一种基因组组装方法,包括以下步骤:
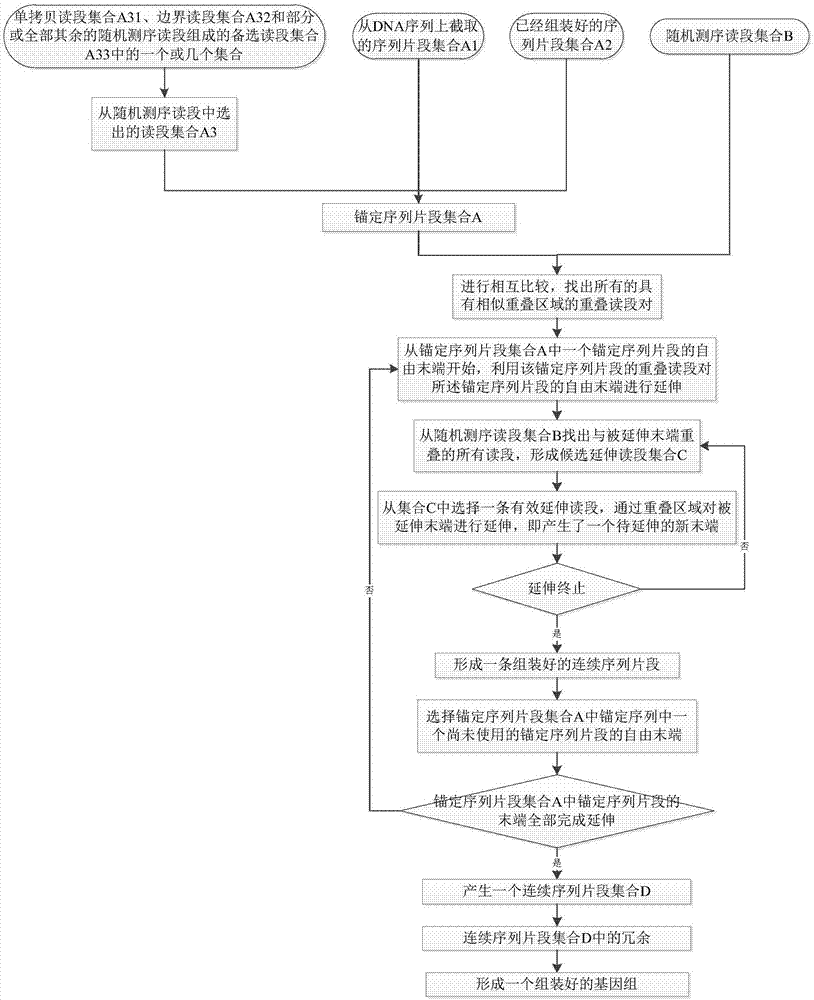
s1,将所有的已知dna序列片段进行相互比较,找出所有的具有相似重叠区域的重叠读段对;其中,所述的已知dna序列包括锚定序列片段集合a和随机测序读段集合b;所述的锚定序列片段集合a包括:从dna序列上截取的序列片段集合a1、已经组装好的序列片段集合a2和从随机测序读段中选出的读段集合a3中的一个或几个集合;所述的将所有的已知dna序列片段进行相互比较,包括将所有的锚定序列片段与所有的测序读段进行相互比较、将所有的测序读段进行相互比较,或者将所有的锚定序列片段进行相互比较;
s2,从锚定序列片段集合a中一个锚定序列片段的自由末端开始,利用该锚定序列片段的重叠读段(即所有的可能读段)对所述锚定序列片段的自由末端进行延伸;延伸时,从随机测序读段集合b找出与被延伸末端重叠的所有读段,形成候选延伸读段集合c,再从集合c中选择一条有效延伸读段,通过重叠区域对被延伸末端进行延伸,即产生了一个待延伸的新末端,重复本步骤直至序列延伸终止,最终形成一条组装好的连续序列片段;
s3,选择锚定序列片段集合a中一个尚未使用的锚定序列片段的自由末端,回到步骤s2,重复步骤s2-s3,直到锚定序列片段集合a中锚定序列片段的末端全部完成延伸(包括延伸不动)为止,最终产生一个连续序列片段集合d;
s4,去除连续序列片段集合d中的冗余,最终形成一个组装好的基因组。
优选的,前述的一种基因组组装方法中,步骤s1中所述的从随机测序读段中选出读段集合a3包括单拷贝读段集合a3、边界读段集合a32和部分或全部其余的随机测序读段组成的备选读段集合a33中的一个或几个集合;其中边界读段包括:(1)位于单拷贝和重复序列的边界上的读段;(2)在重复序列中,其两端跟其它相似的拷贝相比的最高序列一致性差值大于某个阈值的读段,比如一端的序列一致性只有90%(可以当作单拷贝序列来对待),另一端的序列一致性则高达98%,也可以作为边界读段;基于单拷贝序列末端的延伸可以完成绝大部分单拷贝序列的组装,而基于重复序列末端的延伸能够完成大部分重复序列的组装,剩余序列由对备选读段集合中读段的延伸完成;对不同序列末端的延伸可以使用不同的参数,保证了组装效率和质量。
步骤s1中,序列比较多数是以比对的方式来完成,但也有少数情况下,可以不通过比对的方式完成,后者准确度要下降。
优选的,前述的一种基因组组装方法中,单拷贝读段通过以下方法选取:对于每条读段,分别计算其两个末端(比如5kb)的平均覆盖深度,即被重叠读段覆盖的平均次数;若每个读段其两个末端的平均覆盖深度在预设阈值y1(比如平均测序深度的20%~1.5倍)的范围内,则把此读段分成为长度为l(比如100~1000bp),大小一致,重叠长度为l/2的窗口,计算每个窗口的平均覆盖深度;若是没有一个窗口的平均覆盖深度低于预设阈值y2(比如整个读段的平均覆盖深度的20%),则选此读段为有效单拷贝读段;所选的有效单拷贝读段形成单拷贝读段集合a31;此方法保证了选取的是高质量的单拷贝读段,从而保证了测序质量高的单拷贝区域能够被优先组装出来。
优选的,前述的一种基因组组装方法中,边界读段的选取方法如下:对于每条读段,分别计算其两个末端的平均覆盖深度,即被重叠读段覆盖的平均次数;若是有一个末端的平均覆盖深度高于预设阈值y3(比如平均测序深度的1.5倍),而另一个末端的平均覆盖深度不高于预设阈值y3(比如平均测序深度的1.5倍)但不低于预设阈值y4(比如平均测序深度的20%),而且在平均覆盖深度低的一端有多个读段(比如超过25%)具有未比对悬空末端,则此读段被定为边界读段;所有的边界读段形成边界读段集合a32;此方法保证了选取的是高质量的边界读段,从而保证了测序质量高的单拷贝区域和重复区域能够被优先组装出来。
优选的,前述的一种基因组组装方法中,所述步骤s2中序列延伸的终止条件是:没有找到有效重叠读段;或是被延伸末端跟一个终止锚定序列片段的末端有重叠;或是对于从单拷贝序列末端起始的延伸,跟被延伸末端重叠的读段数目少于预设阈值y6或超出了预设阈值y7,或是在跟被延伸末端有重叠的读段中具有悬空末端的条数超出了预设阈值y5(比如所有重叠读段20%的条数);或是对于从重复序列末端起始的延伸,延伸序列的长度超过了预设阈值y8;这些终止条件既保证了单拷贝区域能够被尽可能多且长的组装出来,也保证了重复序列区域组装的完整度,同时也保证了组装序列错误率低,嵌合体少。
优选的,前述的一种基因组组装方法中,所述步骤s2中,在选取锚定序列片段集合a中的读段集合a3中的一个读段作为起始序列时,若是单拷贝读段集合a31或边界读段集合a32不是空集,则备选读段集合a33中的读段不能作为起始序列;选取一个读段作为起始序列时,选取可选集合中最长的一个读段;读段集合a3中的读段不作为终止锚定序列片段;备选读段只是作为最后的补充,保证了基因组组装的完整度,从最长的读段开始延伸,提高了组装效率和质量,不用读段作为终止锚定序列提高了组装序列的连续性。
优选的,前述的一种基因组组装方法中,所述步骤s2中,通过以下方式来从候选延伸读段集合c选择一个有效延伸读段:
s21,根据集合c中每个读段跟被延伸序列片段的比较结果,若是二者(即延伸读段跟被延伸序列片段)的悬空末端长度之和大于预定阈值y9(比如长度超过200bp),则把此读段从集合c中去除;最后将集合c中剩余读段按照重叠区域序列一致性值从高到低排序,然后从序列一致性值最高的读段开始选取,直到从中选取的条数达到了平均测序深度,或是序列一致性值降到了预设阈值y10,选取的读段形成集合c1;这些条件可以保证所选的候选延伸读段基本上都是本地来源的,减少了错误;
s22,根据集合c1中读段相互比较结果,找出具有悬空末端长度之和大于预定阈值y9(比如长度超过200bp)且每对之间都不共享相同读段的所有读段对(即所有的可用于延伸的读段);这样的读段对数若是小于阈值y11(比如集合c1大小20%的对数),把集合c1的读段按照其延伸长度从高到低排序,然后从中选择一条延伸长度排在第三位的读段r为有效延伸读段,以确保延伸末端至少有其他两条读段的支持,以减少错误(当然在任何时候,也都可以选取排在第一位或第二位的读段);这样的读段对数若是大于等于阈值y11,设定集合c中没有有效延伸读段,或者将所有读段按照两两之间在重叠区域的序列一致性是否高于预定阈值y12(比如c1中所有读段之间在重叠区域序列一致性的平均值)分配到同一组,最终形成两组或多组,并从每组之中选取一条延伸长度最长的读段,形成集合c2,再从集合c2中选取一条读段作为有效延伸读段;在选定有效延伸读段r后,将集合c1中的延伸长度不大于所选读段r的延伸长度的所有读段从锚定序列集合a中去除。这些条件保证了最后选取的用于延伸的读段都是高质量的,有其它读段的支持,而在延伸时的候选读段不作为起始读段进行再次延伸,大大减少了计算量,提高了组装效率;这些条件也保证了若是延伸时到达重复序列区域,则还可以从每个拷贝中分别选取一条最好的延伸读段,对不同的拷贝分别进行延伸,从而完成对多个拷贝的分别组装;具体实施时,从延伸读段候选集合c中选择一条延伸读段时,还可以使用延伸读段候选集合c中的其中某一部分序列的共识序列,但是组装速度会变慢。
优选的,前述的一种基因组组装方法中,所述步骤s4中,去除基因组中的冗余的方法是,将连续序列片段集合d中的所有序列片段进行相互比较,找出所有的重叠区域;或者还包括,若是任意两个有重叠的序列片段含有不能比对的悬空末端,则把两个序列都从能够比对到不能比对的边界位置切开;然后去掉跟另一条序列片段相似度高于预设阈值y13且被其完全覆盖的序列片段,连接末端重叠区域超过预设阈值y14的每对序列片段,剩余序列片段则形成一个组装好的基因组;此方法减少了组装序列中的错误率,提高了组装序列的连续性。具体实施时,这个组装好的基因组去掉短的片段后(低于某个阈值),还可以作为锚定序列片段进行进一步的延伸或是连接。
优选的,前述的一种基因组组装方法中,在选择候选延伸读段之前,还包括:设定一个全局序列一致性最低阈值simin;对任一读段来说,首先判断跟其重叠的读段在重叠区域的序列一致性值是否大于等于所述最低阈值simin,如果是,则选用这些重叠读段来作为延伸序列的延伸读段候选,否则放弃选用这些重叠读段来作为延伸序列的延伸读段候选,从而可以去除噪音干扰,提高数据处理的效率和速度,并提高了结果的准确性。
优选的,前述的一种基因组组装方法中,所述的全局序列一致性最低阈值simin参考全基因组水平上的测序读段准确率值α进行设定,比如设定simin=1-(1-α)×3,其中,所述的全基因组水平上的测序读段准确率值α通过以下方式计算获得:取每条读段的具有最高重叠序列一致性的重叠读段,最多取平均测序深度的条数,计算所有重叠区域的平均序列一致性值,作为全基因组水平上的测序读段准确率值α。利用估算出的测序精确度值来设定最低重叠的筛选阈值,可以提高此值设定的准确性,减少背景噪音,提高了结果的准确性和运算速度。
优选的,前述的一种基因组组装方法中,对于末端重叠的两条序列xi和xj(i≠j),其重叠区域的长度分别为:oli_j和olj_i,延伸长度为:eli_j和elj_i,悬空末端的长度为:ohi_j、ohj_i,重叠序列一致性值为:sii_j,重叠分数为:osi_j=sii_j×(oli_j+olj_i)/2-(ohi_j+ohj_i)/2;若是被延伸序列片段xi的重叠末端,设定此末端为右端r,跟延伸序列片段集合{xj}中所有序列片段左端分别有重叠,其中任一j≠i,则xi在重叠末端的序列碱基正确率设为:siir=∑(sii_j×oli_j)/∑oli_j,其中的求和是指对于集合{xj}中的所有j;在一对序列x1、x2的重叠区域,其相同的碱基对数目为m,不同的碱基对数目为mm,x1中插入的碱基数目为i,缺失的碱基数目为d,对于校正后的序列,重叠区域序列一致性值一般设定为:si=m/(m+mm+i+d),若是没有校正的序列,其重叠区域序列一致性值可以设定为si=m/(m+mm),忽略插入和缺失的碱基数目;通过对这些分数的设定和利用,提高了选取正确的序列之间重叠区域的概率。
上述内容中涉及一些术语,为避免混淆,解释如下:
测序读段:测序仪测出的一段dna序列,简称为读段。
连续序列片段:一段连续的dna序列,中间没有缺失的碱基。
覆盖深度:即测序深度,是一个碱基在一组测序读段数据中被测到的次数。
悬空末端:一对有重叠区域的序列,若是在末端有不相似即不能比对的区域,则这些不能比对的末端区域在重叠区域的某一边有两个,其中短的一个末端被称为悬空末端。
共识序列:一条序列作为参考序列跟多条其他序列比对(两两比对或多重序列比对)后,在参考序列的每个碱基位置上取所有序列在此位置上占多数的碱基,最后形成的序列就是共识序列。
自我校正:单分子测序读段碱基错误率高,一个读段跟其相似的多个读段比较后所形成的共识序列可作为这个读段校正后的序列。
嵌合体:两个不在基因组上同一个区域(非相邻,特别是不在同一染色体臂上)的dna序列被连接到了一起,形成一条嵌合体。注意在同一个区域的两个序列,若是因中间缺了一段而连接到了一起,则一般称之为序列缺失,而不称之为嵌合体。
通路:指通过覆瓦式线性重叠形成的dna序列路径,计算长度时需去除序列重叠中的多余部分。
n50长度:就是把序列片段从大到小排序,并对其长度进行累加,当累加长度达到所有序列片段总长度的50%时,最后一个序列片段的长度。其它nx长度,比如n20、n80,以此类推。
发明人经研究发现:序列之间重叠区域的产生,有两种方式:1、来源于基因组上的同一个位置,这些序列的一致性往往很高,但由于测序错误,导致序列的一致性不是100%;2、来源于重复序列的不同拷贝,但这些序列的一致性往往较低;一对重叠序列末端的悬空部分少数是由于测序错误,多数是由于重复序列的不同拷贝造成,因此对跟被延伸序列相比具有悬空末端的读段的处理对于选择正确的延伸读段起到了很重要的作用;选取延伸读段时选择跟被延伸序列相比较序列一致性高的,保证了延伸序列是本地来源的;延伸序列的长度和质量也很重要,在延伸时既考虑了延伸序列的长度,也考虑了延伸序列的质量,帮助优先选择长且质量高的读段,以便在后续的延伸中保证延伸序列的质量。
注意由于dna序列是双链互补的,但在计算重叠区域、序列比较或延伸等时,需要用单链,所以这些操作在两条链上都可以产生;通过链的调整,可以把二者统一起来,不会产生冗余或矛盾。
本发明中,一个序列片段有两个末端,每个末端可定义为一段特定长度(比如1-25kb)的序列,那么所述末端对应的一段特定长度(比如1-25kb)的序列即为末端序列。实际操作中,可通过序列比对的方式去除相似的末端序列(比如一致性>98%),序列缩短后产生新的可用的末端。
与现有技术相比,本发明具有以下优点:
(1)本发明能够从头开始快速高效地完成全基因组的组装,从操作上将全基因组的组装分成两种类型序列的分别组装,第一类,组装单拷贝序列,通过步骤s2和s3,即从单拷贝末端起始的延伸(实际上也包括部分重复序列,特别是被单个测序读段跨过的重复序列),第二类,组装剩余的序列,通过步骤s2和s3,即从非单拷贝读段末端及备选读段末端起始的延伸(后者也包括部分单拷贝序列和部分重复序列)。因为单拷贝序列产生的读段之间的连接关系简单,处理起来简单,也不容易犯错误;而第一类型序列组装完成后,基因组的大部分序列都已完成组装,对剩余序列的组装是一个加强和改进,因而可以简化对整个基因组组装的实施过程,从而使整个方法变得快速高效,不易犯错。而且由于第二类序列组装的存在,能够大大提高组装序列片段的连续性,从而也提高了组装质量。
本发明步骤s1通过将所有的测序读段进行相互比较,找出每对读段之间相似的重叠区域,然后步骤s2-s3选取有效的包含至少一个单拷贝末端的读段,通过延伸,完成单拷贝区域的组装,然后步骤s2-s3通过对剩余末端(包括非单拷贝读段末端和部分可靠性较差的备用读段末端)的延伸,完成对全基因组序列的组装,并通过步骤s4去除冗余和错误,形成一个组装完成的基因组;通过利用本发明的方法构组装全基因组序列,快速高效,也更有利于复原整条染色体及整个基因组的序列。
(2)本发明将随机读段直接连接成通路序列,从而大大提高了基因组组装的效率。而现有技术进行了读段水平上的处理,将相似的读段进行了压缩,在压缩时没有有效区分单拷贝和重复序列,导致处理时效率低,从而多花费了很多时间,而且使得很多来源于不同重复区域的读段被压缩成了一个,这样本来有差别的一些重复区域不能分开;而且由于种种原因,比如测序错误、校正错误等,读段水平上的错误也很容易导致压缩错误,从而降低了基因组组装的效率。
(3)本发明通过设置序列一致性值或覆盖深度或悬空末端的条数或延伸长度,从而根据这些值得设定实现了将基因组中单拷贝序列和重复序列及来源于同一个重复序列家庭的不同重复序列拷贝(各个重复序列的相似度大于90%,特别是相似度>97%)的读段尽可能多地分开,首先将单拷贝读段组装成一个个独立的连续序列片段,然后将每个重复序列来源的读段组装成一个独立的连续序列片段,操作简单,因而将软件编程实施过程大大简化,并提高了运算速度,提高了基因组组装的效率和准确率。
(5)本发明的基因组组装方法还可以用于基因组序列中空白区域的序列填充(将空白区域两端的序列作为锚定序列片段,通过本发明的方法来组装其间的缺失序列),特别是通过结合基因组光学图谱信息或是染色体分组排序信息,组装效果还会大大提高;通过本发明得到的基因组序列片段,可以作为锚定序列片段,进行剩余区域的进一步组装,并通过改变参数来进行多轮循环,控制组装序列的长度,从而进一步提高组装序列的连续性。
(6)本发明的基因组组装方法,可以实现重复序列区域的基因组组装,也可以实现单拷贝序列区域的组装。
(7)本发明的这个方法还可以用于判断任意两个序列之间是否有连接,或是估算两个相邻序列之间的距离。
为了验证本发明的效果,发明人还利用本发明的方案对水稻r498、玉米b73、苦荞pinku1和人hx1的基因组分别进行了基因组组装试验,通过第一步单拷贝序列组装的结果是水稻基因组大小435mb,contign501.12mb;玉米基因组大小2.43gb,contign50216kb,苦荞基因组大小465mb,contign501.65mb;人基因组大小3.43gb,contign504.65mb。进一步对重复序列(第一步遗留的空白)区域进行连接,contign50分别提高到了:6.54mb,13.7mb,21.5mb,16.48mb。这些组装序列的连续性远远高于利用其它现有软件得到的组装结果(r498:canu,1.31mb;b73:pbcr,1.28mb;pinku1:canu,1.1mb;hx1:falcon,8.33mb)。最后组装的基因组大小去掉了冗余,因而更接近于参考基因组的大小。利用本发明组装的水稻基因组中有21个带有端粒序列的contig(10个aaaccct,11个agggttt)。利用本发明组装的玉米基因组中有25个带有端粒序列的contig(13个aaaccct,12个agggttt)。从用时上来说,完成序列校正后,本发明所用时间绝大部分用在了第一步的bwa序列比较上,比如r498,机时总数是2641核小时,而第一步序列比较bwa机时用量是2515核小时。作为比较,现有软件canu的机时总数一般是bwa的3倍以上。如果采用minimap2进行序列比较,r498数据比较机时消耗数降为36核小时。
通过以上试验说明了:(1)本发明的基因组组装方法可以快速构建更长的连续dna序列;(2)本发明可以用于对基因组序列中的空白区域进行序列填充;(3)本发明的方法可以组装重复序列区域;(4)本发明的基因组组装方法的组装速度快,效率更高。
附图说明
图1是本发明的一种实施例的方法流程图;
图2是两个非包含的重叠序列示意图,两端非重叠部分可以相互延伸;ol,重叠序列片段;oh,末端悬空序列片段;el,延伸序列片段;注意oh和el是相对的,二者之间因不能比对,长的片段被认为是el,短的序列片段被称为oh;根据使用场景的不同,比如在判断一个序列是否为潜在的嵌合体时,两个延伸序列el1、el2都可被认为是末端悬空序列片段;
图3是判断一个读段是否为单拷贝-重复序列边界读段的示意图;中间粗线为目标读段,其两端能够跟多个读段重叠,但跟其重叠的读段中有很多在其一端有未比对悬空末端,说明这是一个边界读段;
图4是对一个单拷贝序列利用重叠序列向两端延伸的示意图;
图5是对一个边界序列利用重叠序列进行单端延伸的示意图;
图6是从一个锚定序列延伸到另一个锚定序列的示意图;由重叠的读段组成一条通路;
图7是来源于不同区域的重复序列拷贝的示意图;基因组上的两个相似的重复序列片段r1、r2,跟其比对的上半部分序列是本地产生的测序读段,序列一致性很高,而跟其比对的下半部分序列是另一个重复序列拷贝来源的测序读段,带有悬空末端和碱基变异;
图8是图7对应的重叠图示意图;c1-c4是已知锚定序列片段;r1、r2是重复序列;每个序列有两个末端;u:单拷贝序列,ur,单拷贝和重复区域的边界序列;
图9是测序读段比对后的相互关系示意图;单拷贝序列(u)可以通过局部延伸进行组装;重复序列(r)也可以通过延伸进行组装。
具体实施方式
下面结合附图和具体实施方式对本发明作进一步的说明。
本发明的实施例,一种基因组组装方法,如图1所示,包括以下步骤:
s1,将所有的已知dna序列片段进行相互比较,找出所有的具有相似重叠区域的重叠读段对;其中,所述的已知dna序列包括锚定序列片段集合a和随机测序读段集合b;所述的锚定序列片段集合a包括:从dna序列上截取的序列片段集合a1、已经组装好的序列片段集合a2和从随机测序读段中选出的读段集合a3中的一个或几个集合;所述的将所有的已知dna序列片段进行相互比较,包括将所有的锚定序列片段与所有的测序读段进行相互比较、将所有的测序读段进行相互比较,或者将所有的锚定序列片段进行相互比较;
s2,从锚定序列片段集合a中一个锚定序列片段的自由末端开始,利用该锚定序列片段的重叠读段(即所有的可能读段)对所述锚定序列片段的自由末端进行延伸;延伸时,从随机测序读段集合b找出与被延伸末端重叠的所有读段,形成候选延伸读段集合c,再从集合c中选择一条有效延伸读段,通过重叠区域对被延伸末端进行延伸,即产生了一个待延伸的新末端,重复本步骤直至序列延伸终止,最终形成一条组装好的连续序列片段;
s3,选择锚定序列片段集合a中一个尚未使用的锚定序列片段的自由末端,回到步骤s2,重复步骤s2-s3,直到锚定序列片段集合a中锚定序列片段的末端全部完成延伸(包括延伸不动)为止,最终产生一个连续序列片段集合d;
s4,去除连续序列片段集合d中的冗余,最终形成一个组装好的基因组。
优选的,前述的一种基因组组装方法中,步骤s1中所述的从随机测序读段中选出读段集合a3包括单拷贝读段集合a31、边界读段集合a32和部分或全部其余的随机测序读段组成的备选读段集合a33中的一个或几个集合;其中边界读段包括:(1)位于单拷贝和重复序列的边界上的读段;(2)在重复序列中,其两端跟其它相似的拷贝相比的序列一致性差值大于某个阈值的读段,比如一端的序列一致性只有90%,另一端的序列一致性则高达98%,也可以作为边界读段。
步骤s1中,序列比较多数是以比对的方式来完成,优选采用bwa软件(lih.anddurbinr.,bioinformatics,(2009)25:1754-60.)或是minimap2(lih.,bioinformatics,2018,bty191,)软件,但也有少数情况下,可以不通过比对的方式完成,比如用mhap(https://mhap.readthedocs.io/en/latest/)方法,或是minimap2中的非比对方法,非比对方式的准确度要下降。
步骤s1中,选取单拷贝读段和/或位于单拷贝和重复序列的边界上的边界读段可采取如下方法:
单拷贝读段通过以下方法选取:对于每条读段,分别计算其两个末端(比如5kb)的平均覆盖深度,若每个读段其两个末端的平均覆盖深度在预设阈值y1(比如平均测序深度的20%~1.5倍)的范围内,则把此读段分成为长度为l(比如100~1000bp),大小一致,重叠长度为l/2的窗口,计算每个窗口的覆盖深度;若是没有一个窗口的覆盖深度低于预设阈值y2(比如平均覆盖深度的20%),则选此读段为有效单拷贝读段;所选的有效单拷贝读段形成单拷贝读段集合a31。
边界读段的选取方法如下:把所有读段从长到短排序,对于每条读段,分别计算其两个末端的平均覆盖深度;若是有一个末端的平均覆盖深度高于预设阈值y3(比如平均测序深度的1.5倍),而另一个末端的平均覆盖深度低于预设阈值y3(比如平均测序深度的1.5倍)但不低于预设阈值y4(比如平均测序深度的20%),而且在平均覆盖深度低的一端有多个读段(比如超过25%)具有未比对悬空末端,则此读段被定为边界读段;所有的边界读段形成边界读段集合a32。
上述步骤s2中若起始序列是一个单拷贝读段,则对两个末端都要进行延伸,若起始读段是一个边界读段,则先对对单拷贝末端进行延伸,在所有的单拷贝末端延伸完成后再对重复序列末端进行延伸。在对序列进行延伸时,为增加最后结果正确的可能性,在每一步都只选择很高或最高质量的读段作为候选序列;在上述步骤s2中,对单拷贝序列末端进行延伸时,每一步(除最后一步)选择的读段均为错误率比较低的非边界读段,尽管延伸末端不是最长,但保证了新末端的质量。
上述步骤s2中序列延伸的终止条件可以是:没有找到有效重叠读段;或是被延伸末端跟一个终止锚定序列片段的末端有重叠;或是对于从单拷贝序列末端起始的延伸,跟被延伸末端重叠的读段数目少于预设阈值y6或超出了预设阈值y7,或是在跟被延伸末端有重叠的读段中具有悬空末端的条数超出了预设阈值y5(比如所有重叠读段20%的条数);或是对于从重复序列末端起始的延伸,延伸序列的长度超过了预设阈值y8。
上述步骤s2中,在选取锚定序列片段集合a中的读段集合a3中的一个读段作为起始序列时,若是单拷贝读段集合a31或边界读段集合a32不是空集,则备选读段集合a33中的读段不能作为起始序列,在可选集合中总是取最长的一个读段;读段集合a3中的读段不作为终止锚定序列片段。
上述步骤s2中,通过以下方式来从候选延伸读段集合c选择一个有效延伸读段:
s21,根据集合c中每个读段跟被延伸序列片段的比较结果,若是二者(即延伸读段跟被延伸序列片段)的悬空末端长度之和大于预定阈值y9(比如长度超过200bp),则把此读段从集合c中去除;最后将集合c中剩余读段按照重叠区域序列一致性值从高到低排序,然后从序列一致性值最高的读段开始选取,直到从中选取的条数达到了平均测序深度,或是序列一致性值降到了预设阈值y10,选取的读段形成集合c1;
s22,根据集合c1中读段相互比较结果,找出具有悬空末端长度之和大于预定阈值y9(比如长度超过200bp)且每对之间都不共享相同读段的所有读段对(即所有的可用于延伸的读段);这样的读段对数若是小于阈值y11(比如集合c1大小20%的对数),把集合c1的读段按照其延伸长度从高到低排序,然后从中选择一条延伸长度排在第三位的读段r为有效延伸读段,以确保延伸末端至少有其他两条读段的支持,以减少错误(当然在任何时候,也都可以选取排在第一位或第二位的读段);这样的读段对数若是大于等于阈值y11,设定集合c中没有有效延伸读段,或者将所有读段按照两两之间在重叠区域的序列一致性是否高于预定阈值y12(比如c1中所有读段之间在重叠区域序列一致性的平均值)分配到同一组,最终形成两组或多组,并从每组之中选取一条延伸长度最长的读段,形成集合c2,再从集合c2中选取一条读段作为有效延伸读段;在选定有效延伸读段r后,将集合c1中的延伸长度不大于所选读段r的延伸长度的所有读段从锚定序列集合a中去除。
上述步骤s4中,去除基因组中的冗余的方法是,将连续序列片段集合d中的所有序列片段进行相互比较,找出所有的重叠区域;或者还包括,若是任意两个有重叠的序列片段含有不能比对的悬空末端,则把两个序列都从能够比对到不能比对的边界位置切开;然后去掉跟另一条序列片段相似度高于预设阈值y13且被其完全覆盖的序列片段,连接末端重叠区域超过预设阈值y14的每对序列片段,剩余序列片段则形成一个组装好的基因组。具体实施时,这个组装好的基因组去掉短的片段后(低于某个阈值),就可以作为锚定序列片段进行新的延伸或是利用其他的方法进行连接。
为了去除噪音干扰,提高数据处理的效率和速度,并提高了结果的准确性,可以在选择延伸候选读段之前,还包括:设定一个全局序列一致性最低阈值simin;对任一读段来说,首先判断跟其重叠的读段在重叠区域的序列一致性值是否大于等于所述最低阈值simin,如果是,则选用这些重叠读段来作为延伸序列的延伸读段候选,否则放弃选用这些重叠读段来作为延伸序列的延伸读段候选。
上述的全局序列一致性最低阈值simin参考全基因组水平上的测序读段准确率值α进行设定,比如设定simin=1-(1-α)×3,其中,所述的全基因组水平上的测序读段准确率值α通过以下方式计算获得:取每条读段的具有最高重叠序列一致性的重叠读段,最多取平均测序深度的条数,计算所有重叠区域的平均序列一致性值,作为全基因组水平上的测序读段准确率值α。利用估算出的测序精确度值来设定最低重叠的筛选阈值,可以提高此值设定的准确性,减少背景噪音,提高了结果的准确性和运算速度。
具体实施时,所述的全局序列相似性最低阈值simin也可凭经验设定设定。比如,采用校正后的随机读段作为延伸读段,采用组装好的连续序列片段作为锚定序列。因此在实施时,可采用一个固定的simin值97%,效果已足够好。因为一般校正后的随机测序读段的准确率在99%左右。
具体实施时,对于延伸,也可以只考虑单条重叠的读段,比如选择序列一致性最高的,或延伸长度最长的,而不考虑其它重叠序列的共同影响,但组装效果一般来说要差一些。
对于末端重叠的两条序列xi和xj(i≠j),其重叠区域的长度分别为:oli_j和olj_i,延伸长度为:eli_j和elj_i,悬空末端的长度为:ohi_j、ohj_i,重叠序列一致性值为:sii_j,重叠分数为:osi_j=sii_j×(oli_j+olj_i)/2-(ohi_j+ohj_i)/2;若是被延伸序列片段xi的重叠末端,设定此末端为右端r,跟延伸序列片段集合{xj}中所有序列片段左端分别有重叠,其中任一j≠i,则xi在重叠末端的序列碱基正确率设为:siir=∑(sii_j×oli_j)/∑oli_j,其中的求和是指对于集合{xj}中的所有j;在一对序列x1、x2的重叠区域,其相同的碱基对数目为m,不同的碱基对数目为mm,x1中插入的碱基数目为i,缺失的碱基数目为d,对于校正后的序列,重叠区域序列一致性值一般设定为:si=m/(m+mm+i+d),若是没有校正的序列,其重叠区域序列一致性值可以设定为si=m/(m+mm),忽略插入和缺失的碱基数目。
本实施例是将所有的测序读段进行互相比较,找出每对读段之间相似的重叠区域;为了提高读段的准确率,可以先对该读段进行校正;也可以不进行校正,直接采用原始的随机测序读段进行序列延伸;特别是对单拷贝读段(包括两端都是单拷贝序列的读段)的组装,不需要先对读段进行校正;校正的方法包括用其他测序平台得到的测序错误率很低的测序读段来校正,也包括利用这个集合中的其他的读段来进行自我校正;根据读段之间的重叠关系,选择一个有效单拷贝读段集合和/或一个有效边界读段集合,剩余读段形成一个备用读段集合,以免漏掉基因组中的某些序列;并通过对单拷贝序列末端的延伸,组装基因组中全部或部分单拷贝序列,形成一个连续序列片段集合;再通过对重复序列及备用读段末端的延伸,完成全部的基因组组装,最后通过去除错误和冗余,形成一个组装好的基因组,从而最终完成全基因组序列的组装。
具体实施时,可以构建一个重叠图,是由代表测序读段的节点和他们两两之间的序列重叠作为边构建的无向简单图。每条读段用两个节点来表示,每个节点代表了一个读段末端,而这两个节点之间由一条无向边(在此称之为耦合边)连接;在这个重叠图中,若是两个节点之间有非耦合边的连接,则说明此两个末端之间有重叠,其中的一个可以用来延伸另一个;在遍历图中的通路时,有一个基本要求:在任何时候,进入了一个节点,则必须通过这个节点的耦合边出来(即到达一条已知序列的一个末端节点以后,不能从连接到同一末端节点的其他序列的末端节点出来,而必须从同一序列的另一个末端节点出来,以保证序列的线性延伸);在重叠图中,识别两个序列的两个末端之间是不是有连接可以通过深度搜索或广度搜索来实现;在延伸序列片段时,用到的读段可以从重叠图中去除,减少图的复杂度,并记录形成的通路,代表了组装后的序列,形成的连续序列片段末端也可以直接在图中记录,用于延伸组装剩余的未组装区域。
本发明的一种实施例的工作原理:
如图4、图5所示,两个方法都可以完成部分基因组(重点是单拷贝序列)的组装。图4中从一个单拷贝读段开始向两边延伸,完成一个单拷贝区域的组装;图5显示从一个边界序列的单拷贝端开始,完成一个单拷贝序列的组装。图6是从一个锚定序列延伸到另一个锚定序列的示意图,由重叠的读段组成的一条通路。
如图7(基因组上的两个重复序列片段r1、r2序列一致性很高,但还是有些碱基差异导致他们不完全一致;上半部分比对的序列是本地产生的测序读段,跟所比较的序列之间的差异很小;下半部分比对的序列带有悬空末端,是不同的重复序列拷贝来源的测序读段,跟所比较的序列之间差异较大)、图8(一个重叠图(部分)举例,对应了图7中的序列区域及测序read;c1-c4是锚定序列片段;r1、r2是重复序列;每个序列有两个末端;u:单拷贝序列,ur,单拷贝和重复区域的边界序列)所示,来源于不同区域的重复序列拷贝(所述的重复序列拷贝为相似的序列,属于同一个重复序列家庭)是有差别的。本发明可以实现将来源于上述有差异的重复序列拷贝的读段分别进行组装,形成c1到c2的序列连续片段,及c3到c4的序列连续片段从而完成对整个或部分基因组的组装。
如图9所示,每个区域产生的读段在比对后形成一组相关的序列,通过对其中一些序列作为起始序列进行的延伸,得到这个区域的组装序列;不同的延伸可以得到类似或多余的结果,因而可能产生冗余,需要最后把冗余尽量去除。
本发明的基因组组装方法和现有的sg组装方法之间最关键的区别是本发明把组装单拷贝序列的过程简化处理,在这个过程中不考虑重复序列的组装,因此可以将单拷贝区域通过直接延伸而完成组装;而在重复序列区域,也是通过区分本地来源的读段和不同重复序列拷贝来源的读段而通过延伸来完成整个重复序列区域组装;而sg方法是把重复区域进行了读段水平上的处理,相似的读段进行了压缩,不同的重复区域被压成了一个,这样本来有差别的重复区域不能分开。因为种种原因,比如测序错误,校正错误,等等,读段水平上的错误很容易导致压缩错误;读段之间的重叠已包含了可能的序列上的差异。在延伸时,不同重复拷贝来源的读段不容易连接到一起。反过来说,若是在阈值之上,他们还连接到了一起,则说明这些读段无法区分,这两个区域就会在连接时产生冲突,导致附近重叠序列中有多条有悬空末端,从而产生多种不同的延伸方式;这时,根据方法具体实施的设定,对这个区域的组装过程会自动终止,或是对延伸序列进行分组,对每个组分别进行延伸,直到延伸的长度达到一个预定阈值,或是到达一个终止锚定序列片段末端,从而完成对这个区域的序列组装。
在实践中,由于序列之间的差异不是完全均匀分布的,或是由于测序错误没有得到全部校正,会导致误差,但总的来说,绝大多数<99%的重复序列拷贝是很容易区分的。目前单分子测序产生的原始读段的平均错误率在10%-15%。通过自我校正后的读段平均错误率大大降低,比如很多读段的错误率能降低到1%以下。单拷贝序列即使未做校正,也很容易区分;而对于绝大多数基因组上相似度小于98%的重复序列来说,其产生的读段在校正后相似度都不会高于98%(少数情况下由于校正错误,会产生更相似的序列),因而在序列比对及组装的过程中都很容易区分。
- 还没有人留言评论。精彩留言会获得点赞!