一种核苷四磷酸的合成方法与流程
本发明涉及化学合成和生物化学领域,具体涉及一种核苷四磷酸的合成方法。
背景技术:
对核苷多磷酸的探索一直是人们非常感兴趣的课题,它们在分子生物技术,生物学研究和细胞遗传学等方面有广泛的应用。核苷多磷酸盐(nps)及其结合物在许多生物过程中发挥关键作用。核苷三磷酸(ntps)是dna和rna构建模块的前体,它们启动信号通路(即gtp),而三磷酸腺苷atp是许多生命系统中的关键能量来源。核苷二磷酸糖(ndp-糖)在多糖的酶促合成中起到糖基供体的作用。内源性二核苷五磷酸(包括ap5a,ap5g和gp5g)是必需的细胞外信号分子,在心血管和神经系统的生理和病理过程中发挥重要作用。ap5a和ap5g表现出对血管平滑肌细胞(vsmcs)的血管调节作用和增殖作用,而gp5g诱导vsmcs增殖。除了它们的天然生物学作用外,核苷多磷酸及其结合物已被广泛用作治疗性重要酶的抑制剂和探针,而up4u二核苷酸最近已被批准用作药物。含有2'-脱氧核苷-5'-四磷酸的末端荧光标记化合物可作为dna聚合酶的多功能底物。这些修饰的四磷酸盐已被用于各种分子生物学应用,例如dna标记和测序,snp分析,核酸扩增和pcr。末端嘧啶四磷酸衍生物已被用作选择性激动剂p2y4受体,而制备含2'-脱氧核苷-5'-四磷酸的末端荧光标记的前体是2'-脱氧核苷-5'-四磷酸,所以合成多磷酸核苷的重要性不言而喻。除了2'-脱氧核苷-5'-四磷酸外,核糖核苷-5'-四磷酸也参与各种分子生物学应用。例如,天然核苷酸腺苷-5'-四磷酸存在于许多生物系统中,并且通过p2x受体起到眼内压的生理调节剂的作用。腺苷-5'-四磷酸已被确定为最有效的嘌呤能内皮源性血管收缩剂。鸟苷-5'-四磷酸和腺苷-5'-四磷酸作为酵母酿酒酵母细胞质外多磷酸酶的底物。
目前,用于制备核苷多磷酸的合成方法仍然非常有限,并且其大部分方法均需要将2'-或者-3'-oh保护起来,很难做到不需要保护即能一锅法高选择性地生成5'-核苷四磷酸。korear使用一锅法合成2'-脱氧腺苷-3'-o-四磷酸(2'-d-3'-a4p)和2'-脱氧胞苷-3'-o-四磷酸(2'-d-3'-c4p)。通过使用一锅法合成将n6-苯甲酰基-5'-o-乙酰丙酰基-2'-脱氧腺苷转化成n6-苯甲酰基-5'-o-乙酰丙酰基-2'-脱氧腺苷-3'-o-四磷酸酯,但是其磷酸化试剂选择性很弱。后来又以相同的合成策略成功地开发了2'-脱氧胞苷-5'-四磷酸(dc4p)和胸苷-5'-四磷酸(t4p)的有效化学合成。合成需要三个步骤,涉及到使用三氯氧化磷使2'-脱氧核苷单磷酸化,将5'-单磷酸转化为相应的咪唑盐,接着与三[三丁基铵]三磷酸反应产生2'-脱氧核苷-5'-四磷酸。mohamadys等研究人员开发了合成二核苷5’-五磷酸(np5n)和核苷5’-四磷酸(np4)的方法。该过程依赖于环状三偏磷酸盐的活化,接着与核苷5’-单磷酸酯(nmp)反应以产生中间体,中间体与水或nmp反应得到所需产物,产率为77-86%。也有文献报道核苷三磷酸与dcc反应得到环状三偏磷酸酯,随后用正磷酸酯开环以提供相应的核苷四磷酸。korear还在文献中说明脱氧核苷的高选择性单磷酸化取决于碱的正确选择。2-脱氧嘌呤核苷的最优碱是三丁胺,而在2-脱氧-胸苷的情况下,质子海绵则是合适的碱,2-脱氧胞苷则不需要碱的参与。
由于核苷多磷酸应用的多样性以及它们在生物过程中发挥的关键作用,核苷多磷酸盐的合成一直是研究的热点,新颖、高效的合成方法对于核酸化学是非常有价值的。文献已报道的合成方法主要有:(1)运用高度活泼地亚磷酸化剂反应,因为核苷上多种官能团(伯羟基和仲羟基和氨基)的存在,这样就需要保护和去保护这些官能团以减少不需要的副产物。由于核苷的5’-羟基比3’-和2’-羟基更有活性,保护2’-羟基以及核碱基上的氨基是比较麻烦的。(2)使用高活性磷酰氯(pocl3)作为磷酸化剂,并且在焦磷酸盐处理之前使用三甲基磷酸酯溶剂来降低pocl3反应性,主要在5’-位(相当于活化的单磷酸酯)产生二氯磷酸酯。理论上,这样就不需要对核苷进行保护。然而实际上,这种策略通常会产生大量副产物(如区域异构体和低聚磷酸盐),并导致纯化困难,最后可能产率很低。(3)通过运用酶的方法制备核苷多磷酸也受限于酶的种类和底物特异性。
技术实现要素:
本发明要解决的技术问题是,针对现有技术不足,提供一种核苷四磷酸的合成方法。该方法所需原料简单、便宜,反应条件温和,操作简单,可适合大规模生产。
本发明设计并开发了一种核苷5’-四磷酸的一锅法合成方法,在此过程中对核苷无需任何保护。通过原位生成的亚磷酸化试剂与未保护的核苷的5’-羟基基团选择性反应,从而实现简单高效地合成核苷5’-四磷酸。为了减少不需要的副产物并获得高质量的产品,我们选用了温和的、高选择性的磷酸化试剂。合成的四磷酸质量高,容易纯化,可以通过dna聚合酶有效地参与dna链的延伸。这种新颖的合成方法对于四磷酸来说是最直接且经济的。
本发明的目的是通过以下技术方案实现的:
第一方面,本发明提供了一种核苷四磷酸的合成方法,包括以下步骤:
将核苷与环状磷酸化试剂发生选择性磷酸化反应,再经氧化、水解开环,即得所述核苷四磷酸。
优选地,所述环状磷酸化试剂的结构如式i所示:
优选地,所述环状磷酸化试剂的制备方法包括:
将
优选地,所述
优选地,所述
优选地,所述核苷包括脱氧核糖核苷、核糖核苷中的一种。
优选地,所述核苷与环状磷酸化试剂发生选择性磷酸化反应所得产物中间体的结构如式ii所示:
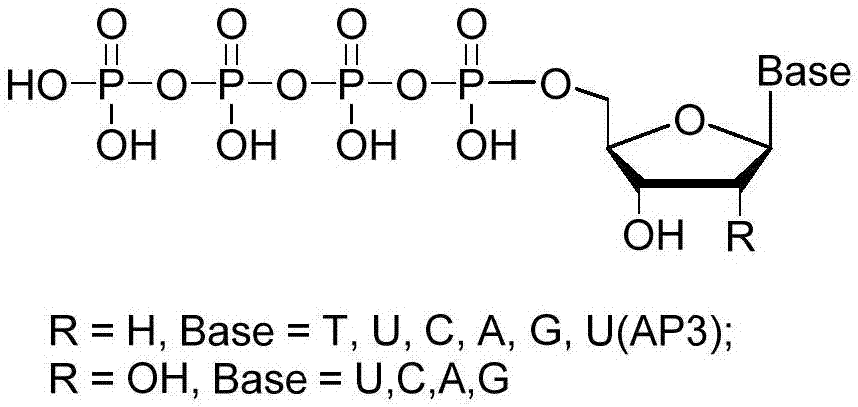
其中,r为h或oh;base=t,u,c,a,g,u(ap3)。
优选地,所述氧化、水解开环的具体步骤如下:
将核苷与原位生成的环状磷酸化试剂发生选择性磷酸化反应所得产物中间体与碘溶液反应,即可。
优选地,所述碘溶液的质量浓度为1-5%;所述碘溶液中的溶剂为体积比1:1-12:1的py和h2o的混合溶液。
本发明合成核苷四磷酸的反应机理是:所述反应过程中原位生成环状磷酸化试剂作为重要反应中间体,该反应中间体反应活性较弱,但是它能够选择性地在5'-oh位置生成四磷酸,而在3'-oh以及2'-oh接上四磷酸的可能性很小,从而不需要保护3'-oh以及2'-oh,最终能够一锅法得到我们需要的5'核苷四磷酸。
本发明制备的核苷四磷酸,使用荧光素标记生成荧光标记的核苷四磷酸,可进一步用于基因测序。
与现有技术相比,本发明具有如下的有益效果:
1.本发明提供了一种核苷四磷酸的合成方法,该方法用于合成核苷四磷酸的过程中,应用原位生成的高选择性磷酸化试剂,选择性地在核苷的5'位羟基接四磷酸,而有效抑制了3’(或者2’-)羟基的磷酸化,能够选择性地生成高纯度5’-核苷四磷酸。并且在此过程中不需要将3’(或者2’-)羟基保护起来,实现了真正意义上的一锅法且不需保护的核苷四磷酸合成方法,该方法既适用于脱氧核糖核苷四磷酸的合成,也适用于核糖核苷四磷酸的合成。
2.合成的核苷四磷酸可有效地被dna聚合酶识别,并参与dna链的延伸,进一步在核苷四磷酸的末端接荧光素,形成的荧光标记核苷四磷酸可用于基因测序。
3.核苷四磷酸在dna测序、标记、延伸等生物学领域广泛使用,目前其销售价格很高,且合成方法复杂,难以控制。本发明的合成方法所需原料简单易得,合成过程均为常规化学反应,可用于大规模推广使用。
附图说明
通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
图1为核苷四磷酸总的结构式;
图2为核苷四磷酸的合成路线图;
图3为实施例1所述脱氧核糖胸腺嘧啶核苷四磷酸dt4p的hplc谱图;
图4为实施例1所述dt4p的质谱;
图5为实施例1所述dt4p的1h-nmr谱图;
图6为实施例1所述dt4p的31p-nmr谱图;
图7为实施例3所述脱氧核糖胞嘧啶核苷四磷酸dc4p的31p-nmr谱图;
图8为实施例3所述dc4p的hplc谱图;
图9为实施例7所述核糖胞嘧啶核苷四磷酸c4p的hplc谱图;
图10为实施例7所述c4p的31p-nmr谱图。
具体实施方式
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变化和改进。这些都属于本发明的保护范围。
实施例1
本实施例核苷酸的结构式如式(iii)所示(图1):
相应的合成路线如图2所示;具体包括如下步骤:
第一步、(nhbu3)3h2p3o10制备
取23g离子交换树脂(dowex50wx8)装于柱中,称取1g三聚磷酸钠亦装于柱中,用去离子水慢慢洗涤,洗涤液用圆底烧瓶接,圆底烧瓶置于冰浴中,圆底烧瓶内含1ml三正丁胺和10ml乙醇。待流出液的ph慢慢接近中性时,停止洗涤。将所得混合溶液旋干,加入乙醇再次旋干,重复两次,加入无水dmf,再次旋干,得黄色粘稠状物质,加入分子筛与无水dmf溶解,保存于干燥器中。
第二步、一锅法制备核苷四磷酸
在手套箱中称取2-氯-4h-1,3,2-苯并二氧磷-4-酮50mg(1eq)于反应管中,用1ml无水dmf溶解,室温下搅拌半小时,得溶液a。抽取(nhbu3)3h2p3o10340mg(2eq)并加入2ml无水dmf,1.5ml无水三正丁胺,室温下搅拌半小时,得溶液b。将溶液a加入到溶液b中,继续搅拌半小时,得溶液c,即生成了化合物i。然后称取2-脱氧胸苷50mg(1eq)于50ml单口瓶中,抽真空并干燥,充入氮气保护,用1ml无水dmf将其溶解,得溶液d。将溶液c加入到溶液d中,室温搅拌1.5h,生成化合物ii;然后加入4ml3%的碘溶液(9:1的py:h2o),体系15min内呈深红色不褪色,之后加入5ml水,室温搅拌2h。加入1.5ml的3mol/l氯化钠水溶液与22ml乙醇。将体系中的混合溶液转移入离心管内,置于-80℃中冷冻1.5h,生成沉淀。之后通过3200r/min的20min离心,取下层固体。达到初步纯化的作用。
将固体用去离子水溶解,经过滤后通过制备型hplc分离。hplc分离条件:制备柱规格为21.2*250mm,10μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-80min,0-40%的甲醇,流速为6ml/min,约28min时出峰。
分析型hplc条件:分析柱规格为4.6*250mm,5μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-100min,0-40%的甲醇,流速为1ml/min,约8min时出峰。所得产物脱氧核糖胸腺嘧啶核苷四磷酸dt4p的hplc谱图如图3所示,质谱图如图4所示,1h-nmr谱图如图5所示,31p-nmr谱图如图6所示。1hnmr(400mhz,d2o)δ7.56(d,j=6.5hz,1h),6.11(dd,j=8.9,4.6hz,1h),4.42(d,j=14.7hz,1h),3.97(d,j=17.7hz,3h),2.19–2.08(m,2h),1.71(s,3h);31pnmr(162mhz,d2o)δ-8.27(d,j=19.8hz,1p),-9.09(dd,j=36.8,19.8hz,1p),-20.11–-21.20(m,2p);ltq(esi-):m/zcalcd.forc10h18n2o17p4[m-h]561.15;found561.16.
产物dt4p的产率为20%;纯度为99.9%。
核苷四磷酸合成方法的反应机理如下所示:
在该反应过程中,原位生成的式i中间体作为一种高选择性的磷酸化试剂,继续与加入反应体系的核苷反应,经过一种八元环的中间体过渡态,然后氧化、水解后开环,最终得到预期的核苷四磷酸。
上述合成方法得到的核苷四磷酸,经荧光标记后,可用于dna测序。
实施例2
本实施例核苷酸的结构式如式(i)所示:
相应的合成路线如图2所示;具体包括如下步骤:
在手套箱中称取2-氯-4h-1,3,2-苯并二氧磷-4-酮50mg(1eq)于反应管中,用1ml无水dmf溶解,室温下搅拌半小时,得溶液a。抽取(nhbu3)3h2p3o10340mg(2eq)并加入2ml无水dmf,1.5ml无水三正丁胺,室温下搅拌半小时,得溶液b。将溶液a加入到溶液b中,继续搅拌半小时,得溶液c,即生成了化合物i。然后称取2-脱氧尿苷1eq于50ml单口瓶中,抽真空并干燥,充入氮气保护,用1ml无水dmf将其溶解,得溶液d。将上述溶液c加入到溶液d中,室温搅拌1.5h,生成化合物ii。然后加入4ml3%的碘溶液(9:1的py:h2o),体系15min内呈深红色不褪色,之后加入5ml水,室温搅拌2h。加入1.5ml的3mol/l氯化钠水溶液与22ml乙醇。将体系中的混合溶液转移入离心管内,置于-80℃中冷冻1.5h。之后通过3200r/min的20min离心,取下层固体。
将固体用去离子水溶解,经过滤后通过制备型hplc分离。hplc分离条件:制备柱规格为21.2*250mm,10μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-80min,0-40%的甲醇,流速为6ml/min。
分析型hplc条件:分析柱规格为4.6*250mm,5μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-100min,0-40%的甲醇,流速为1ml/min。
hrms(esi-):m/zcalcdforc9h16n2o17p4[m-h]548.1190;found548.1204.
所得产物的产率为12%;纯度为99%。
实施例3
本实施例核苷酸的结构式如式(i)所示:
相应的合成路线如图2所示;具体包括如下步骤:
在手套箱中称取2-氯-4h-1,3,2-苯并二氧磷-4-酮50mg(1eq)于反应管中,用1ml无水dmf溶解,室温下搅拌半小时,得溶液a。抽取(nhbu3)3h2p3o10340mg(2eq)并加入2ml无水dmf,1.5ml无水三正丁胺,室温下搅拌半小时,得溶液b。将溶液a加入到溶液b中,继续搅拌半小时,得溶液c,即生成化合物i。称取1.0eqdc于50ml单口瓶中,抽真空并干燥,充入氮气保护,用1ml无水dmf将其溶解,得溶液d。将上述溶液c加入到溶液d中,室温搅拌1.5h,生成化合物ii。然后加入4ml3%的碘溶液(9:1的py:h2o),体系15min内呈深红色不褪色,之后加入5ml水,室温搅拌2h。加入1.5ml的3mol/l氯化钠水溶液与22ml乙醇。将体系中的混合溶液转移入离心管内,置于-80℃中冷冻1.5h。之后通过3200r/min的20min离心,取下层固体。
将固体用去离子水溶解,经过滤后通过制备型hplc分离。hplc分离条件:制备柱规格为21.2*250mm,10μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-80min,0-40%的甲醇,流速为6ml/min。分析型hplc条件:分析柱规格为4.6*250mm,5μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-100min,0-40%的甲醇,流速为1ml/min。所得产物脱氧核糖胞嘧啶核苷四磷酸dc4p的31p-nmr谱图如图7所示,hplc谱图如图8所示。
1hnmr(400mhz,d2o)δ7.91(d,j=7.6hz,1h),6.08(d,j=7.3hz,1h),5.91(d,j=4.3hz,1h),4.30(s,1h),4.24(t,j=4.7hz,1h),4.17(s,3h),3.94(m,j=7.4hz,1h);31pnmr(162mhz,d2o)δ-10.70(d,j=19.2hz,1p),-11.51(d,j=19.9hz,1p),-23.19(dd,j=41.5,22.9hz,2p);ms(esi-tof):m/zcalcd.forc9h17n3o17p4[m-h]561.9508;found561.9418
所得产物的产率为12%;纯度为99.9%。
实施例4
本实施例核苷酸的结构式如式(i)所示:
相应的合成路线如图2所示;具体包括如下步骤:
在手套箱中称取2-氯-4h-1,3,2-苯并二氧磷-4-酮50mg(1eq)于反应管中,用1ml无水dmf溶解,室温下搅拌半小时,得溶液a。抽取(nhbu3)3h2p3o10340mg(2eq)并加入2ml无水dmf,1.5ml无水三正丁胺,室温下搅拌半小时,得溶液b。将溶液a加入到溶液b中,继续搅拌半小时,得溶液c,即生成化合物i。称取1.0eqda于50ml单口瓶中,抽真空并干燥,充入氮气保护,用1ml无水dmf将其溶解,得溶液d。将上述溶液c加入到溶液d中,室温搅拌1.5h,生成化合物ii;然后加入4ml3%的碘溶液(9:1的py:h2o),体系15min内呈深红色不褪色,之后加入5ml水,室温搅拌2h。加入1.5ml的3mol/l氯化钠水溶液与22ml乙醇。将体系中的混合溶液转移入离心管内,置于-80℃中冷冻1.5h。之后通过3200r/min的20min离心,取下层固体。
将固体用去离子水溶解,经过滤后通过制备型hplc分离。hplc分离条件:制备柱规格为21.2*250mm,10μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-80min,0-40%的甲醇,流速为6ml/min。
分析型hplc条件:分析柱规格为4.6*250mm,5μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-100min,0-40%的甲醇,流速为1ml/min。
hrms(esi-):m/zcalcdforc9h16n2o17p4[m-h]588.9777;found588.9769.
所得产物的产率为15%;纯度为99.4%。
实施例5
本实施例核苷酸的结构式如式(i)所示:
相应的合成路线如图2所示;具体包括如下步骤:
在手套箱中称取2-氯-4h-1,3,2-苯并二氧磷-4-酮50mg(1eq)于反应管中,用1ml无水dmf溶解,室温下搅拌半小时,得溶液a。抽取(nhbu3)3h2p3o10340mg(2eq)并加入2ml无水dmf,1.5ml无水三正丁胺,室温下搅拌半小时,得溶液b。将溶液a加入到溶液b中,继续搅拌半小时,得溶液c,即生成化合物i。称取1.0eqdg于50ml单口瓶中,抽真空并干燥,充入氮气保护,用1ml无水dmf将其溶解,得溶液d。将上述溶液c加入到溶液d中,室温搅拌1.5h,生成化合物ii;然后加入4ml3%的碘溶液(9:1的py:h2o),体系15min内呈深红色不褪色,之后加入5ml水,室温搅拌2h。加入1.5ml的3mol/l氯化钠水溶液与22ml乙醇。将体系中的混合溶液转移入离心管内,置于-80℃中冷冻1.5h。之后通过3200r/min的20min离心,取下层固体。
将固体用去离子水溶解,经过滤后通过制备型hplc分离。hplc分离条件:制备柱规格为21.2*250mm,10μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-80min,0-40%的甲醇,流速为6ml/min。
分析型hplc条件:分析柱规格为4.6*250mm,5μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-100min,0-40%的甲醇,流速为1ml/min。
hrms(esi-):m/zcalcdforc9h16n2o17p4[m-h]586.9621;found586.9616.
所得产物的产率为14%;纯度为99.1%。
实施例6
本实施例核苷酸的结构式如式(i)所示:
相应的合成路线如图2所示;具体包括如下步骤:
在手套箱中称取2-氯-4h-1,3,2-苯并二氧磷-4-酮50mg(1eq)于反应管中,用1ml无水dmf溶解,室温下搅拌半小时,得溶液a。抽取(nhbu3)3h2p3o10340mg(2eq)并加入2ml无水dmf,1.5ml无水三正丁胺,室温下搅拌半小时,得溶液b。将溶液a加入到溶液b中,继续搅拌半小时,得溶液c,即生成化合物i。称取尿苷1eq于50ml单口瓶中,抽真空并干燥,充入氮气保护,用1ml无水dmf将其溶解,得溶液d。将上述溶液c加入到溶液d中,室温搅拌1.5h,生成化合物ii;然后加入4ml3%的碘溶液(py:h2o为9:1),体系15min内呈深红色不褪色,之后加入5ml水,室温搅拌2h。加入1.5ml的3mol/l氯化钠水溶液与22ml乙醇。将体系中的混合溶液转移入离心管内,置于-80℃中冷冻1.5h。之后通过3200r/min的20min离心,取下层固体。
将固体用去离子水溶解,经过滤后通过制备型hplc分离。hplc分离条件:制备柱规格为21.2*250mm,10μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-80min,0-40%的甲醇,流速为6ml/min。
分析型hplc条件:分析柱规格为4.6*250mm,5μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-100min,0-40%的甲醇,流速为1ml/min。
hrms(esi-):m/zcalcdforc9h16n2o17p4[m-h]563.9349;found563.9340.
所得产物的产率为9.3%;纯度为99.9%。
实施例7
本实施例核苷酸的结构式如式(i)所示:
相应的合成路线如图2所示;具体包括如下步骤:
在手套箱中称取2-氯-4h-1,3,2-苯并二氧磷-4-酮50mg(1eq)于反应管中,用1ml无水dmf溶解,室温下搅拌半小时,得溶液a。抽取(nhbu3)3h2p3o10340mg(2eq)并加入2ml无水dmf,1.5ml无水三正丁胺,室温下搅拌半小时,得溶液b。将溶液a加入到溶液b中,继续搅拌半小时,得溶液c,即生成化合物i。称取1.0eq核苷c于50ml单口瓶中,抽真空并干燥,充入氮气保护,用1ml无水dmf将其溶解,得溶液d。将上述溶液c加入到溶液d中,室温搅拌1.5h,生成化合物ii;然后加入4ml3%的碘溶液(9:1的py:h2o),体系15min内呈深红色不褪色,之后加入5ml水,室温搅拌2h。加入1.5ml的3mol/l氯化钠水溶液与22ml乙醇。将体系中的混合溶液转移入离心管内,置于-80℃中冷冻1.5h。之后通过3200r/min的20min离心,取下层固体。
将固体用去离子水溶解,经过滤后通过制备型hplc分离。hplc分离条件:制备柱规格为21.2*250mm,10μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-80min,0-40%的甲醇,流速为6ml/min。分析型hplc条件:分析柱规格为4.6*250mm,5μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-100min,0-40%的甲醇,流速为1ml/min。所得产物核糖胞嘧啶核苷四磷酸c4p的hplc谱图如图9所示,31p-nmr谱图如图10所示。
1hnmr(400mhz,d2o)δ7.89(d,j=7.5hz,1h),6.24(t,j=6.6hz,1h),6.05(d,j=7.5hz,1h),4.53(d,j=2.8hz,1h),4.11(d,j=4.6hz,3h),2.30(dd,j=18.9,15.3hz,1h),2.25–2.16(m,1h);31pnmr(162mhz,d2o)δ-10.54(d,j=19.9hz,1p),-11.50(d,j=19.8hz,1p),-23.33(t,j=19.8hz,2p);hrms(esi-tof):m/zcalcdforc9h17n3o17p4[m-h]561.9508;found561.9418.
所得产物的产率为13%;纯度为99.9%。
实施例8
本实施例核苷酸的结构式如式(i)所示:
相应的合成路线如图2所示;具体包括如下步骤:
在手套箱中称取2-氯-4h-1,3,2-苯并二氧磷-4-酮50mg(1eq)于反应管中,用1ml无水dmf溶解,室温下搅拌半小时,得溶液a。抽取(nhbu3)3h2p3o10340mg(2eq)并加入2ml无水dmf,1.5ml无水三正丁胺,室温下搅拌半小时,得溶液b。将溶液a加入到溶液b中,继续搅拌半小时,得溶液c,即生成化合物i。称取1.0eq核苷a于50ml单口瓶中,抽真空并干燥,充入氮气保护,用1ml无水dmf将其溶解,得溶液d。将上述溶液c加入到溶液d中,室温搅拌1.5h,生成化合物ii;然后加入4ml3%的碘溶液(9:1的py:h2o),体系15min内呈深红色不褪色,之后加入5ml水,室温搅拌2h。加入1.5ml的3mol/l氯化钠水溶液与22ml乙醇。将体系中的混合溶液转移入离心管内,置于-80℃中冷冻1.5h。之后通过3200r/min的20min离心,取下层固体。
将固体用去离子水溶解,经过滤后通过制备型hplc分离。hplc分离条件:制备柱规格为21.2*250mm,10μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-80min,0-40%的甲醇,流速为6ml/min。
分析型hplc条件:分析柱规格为4.6*250mm,5μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-100min,0-40%的甲醇,流速为1ml/min。
hrms(esi-):m/zcalcdforc9h16n2o17p4[m-h]604.9726;found604.9735.
所得产物的产率为16%;纯度为99.4%。
实施例9
本实施例核苷酸的结构式如式(i)所示:
相应的合成路线如图2所示;具体包括如下步骤:
在手套箱中称取2-氯-4h-1,3,2-苯并二氧磷-4-酮50mg(1eq)于反应管中,用1ml无水dmf溶解,室温下搅拌半小时,得溶液a。抽取(nhbu3)3h2p3o10340mg(2eq)并加入2ml无水dmf,1.5ml无水三正丁胺,室温下搅拌半小时,得溶液b。将溶液a加入到溶液b中,继续搅拌半小时,得溶液c,即生成化合物i。称取1.0eq核苷g于50ml单口瓶中,抽真空并干燥,充入氮气保护,用1ml无水dmf将其溶解,得溶液d。将上述溶液c加入到溶液d中,室温搅拌1.5h,生成化合物ii;然后加入4ml3%的碘溶液(9:1的py:h2o),体系15min内呈深红色不褪色,之后加入5ml水,室温搅拌2h。加入1.5ml的3mol/l氯化钠水溶液与22ml乙醇。将体系中的混合溶液转移入离心管内,置于-80℃中冷冻1.5h。之后通过3200r/min的20min离心,取下层固体。
将固体用去离子水溶解,经过滤后通过制备型hplc分离。hplc分离条件:制备柱规格为21.2*250mm,10μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-80min,0-40%的甲醇,流速为6ml/min。
分析型hplc条件:分析柱规格为4.6*250mm,5μm,流动相为20mmteaa缓冲液与甲醇,紫外检测波长为254nm,0-100min,0-40%的甲醇,流速为1ml/min。
hrms(esi-):m/zcalcdforc9h16n2o17p4[m-h]602.9570;found602.9560.
所得产物的产率为12%;纯度为99.9%。
实施例10
本实施例考察了采用
实施例11、对实施例合成的核苷四磷酸参与dna链延伸的验证
实验步骤:将常规碱基互补配对的引物和模板溶于tris-edta缓冲液(te,ph7.5)中,按照以下条件退火:95℃保温3分钟,然后以0.1℃/秒的速率降温至4℃并保温。经过退火的引物和模板形成双链,可用于dna延伸反应。在pcr管中依次加入:2μl10xklenow反应缓冲液,1.5μl氯化钠溶液(1m),1.5μl经过退火的引物和模板(1μg/μl),5μlt4p(1mm),0.4μl(2u)ofklenowfragmentexo-dna聚合酶,9.6μl灭菌水,形成总体积为20μl的反应体系。混合均匀后,将此反应体系在37℃下保温15分钟,然后升温至72℃保温10分钟,最后自然冷却至16℃。经过酚氯仿抽提和乙醇沉淀,得到延伸产物。所述延伸产物用变性胶图验证的确能够参与dna链延伸。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变化或修改,这并不影响本发明的实质内容。在不冲突的情况下,本申请的实施例和实施例中的特征可以任意相互组合。
- 还没有人留言评论。精彩留言会获得点赞!