利伐沙班噁唑烷杂质对照品及其制备方法与流程
本发明涉及药物杂质,具体涉及一种利伐沙班噁唑烷杂质对照品及其制备方法。
背景技术:
利伐沙班(rivaroxaban,结构式如式ii所示)是一种口服、具有生物利用度的xa因子抑制剂,其选择性地阻断xa因子的活性位点,且不需要辅因子(例如抗凝血酶iii)以发挥活性。通过内源性及外源性途径活化x因子为xa因子(fxa),在凝血级联反应中发挥重要作用。
在进行利伐沙班合成过程中,通过杂质分析,发现一种新的噁唑烷杂质,该杂质结构式如式i所示,该杂质可能为利伐沙班的一种降解杂质,目前尚未见公开资料报道过该杂质及其合成。
药品上市前必须进行质量、安全性和效能科学评价,药物杂质与药物质量以及药物安全性密切相关,杂质对照品可帮助确定杂质的合理限度,对药品检验方法及质量标准的建立将会起着极大的促进作用。
技术实现要素:
发明目的:本发明的目的是提供一种利伐沙班噁唑烷杂质对照品;
本发明的另一目的是提供一种工艺简单,产品纯度高的制备该杂质对照品的方法,为利伐沙班的质量控制提供合格的噁唑烷杂质对照品。
技术方案:本发明提供一种利伐沙班噁唑烷杂质,该杂质结构式如式i所示:
本发明另一方面提供上述利伐沙班噁唑烷杂质的制备方法,该制备方法包括以下步骤:
1)将利伐沙班在碱性条件下水解开环,制得5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺;步骤1)反应式如下:
2)在二氯亚砜作用下,使步骤1)制得的5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺关环,制得利伐沙班噁唑烷杂质;步骤2)反应式如上:
优选地,步骤1)中,碱性条件是通过向水解开环的反应体系中加入强碱形成的,使用的利伐沙班与强碱的摩尔比为1∶2~1∶6;强碱可以为氢氧化钠、氢氧化钾或氢氧化锂;水解开环的反应溶剂为水与甲醇形成的混合溶剂或水与乙醇形成的混合溶剂,混合溶剂中,水与甲醇或乙醇的体积比为1∶1~1∶5;水解开环的反应温度为50℃~85℃,反应时间为3~8小时。
步骤2)中,5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺与二氯亚砜的摩尔比为1∶2~1∶5;使用的反应溶剂为二氯甲烷、三氯甲烷、四氯化碳或四氢呋喃,反应温度为25℃~50℃,反应时间为5~8小时。
有益效果:本发明提供的利伐沙班噁唑烷杂质对照品的制备方法工艺简单,纯度高,能够为利伐沙班的质量控制提供合格的噁唑烷杂质对照品。
附图说明
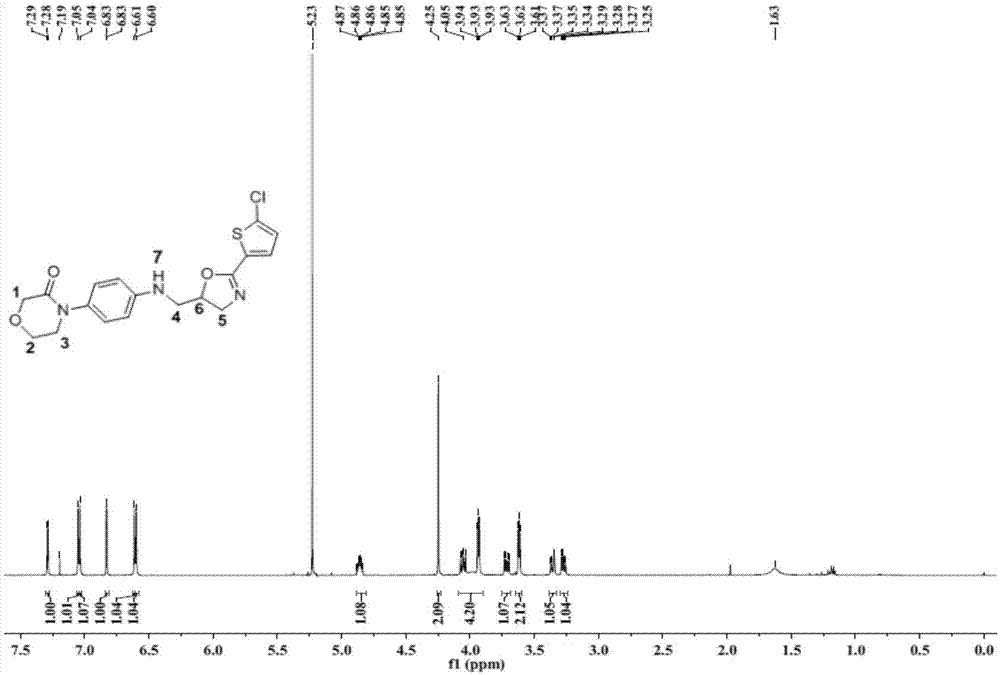
图1是利伐沙班噁唑烷杂质对照品的核磁共振氢谱图;
图2是利伐沙班噁唑烷杂质对照品的质谱图。
具体实施方式
本发明中使用的药品来源如下:
利伐沙班来自南京中邦制药有限公司;
二氯亚砜购买自国药基团化学试剂有限公司。
实施例1
利伐沙班噁唑烷杂质对照品的制备方法如下:
1)在500ml反应瓶中依次加入10.0g利伐沙班固体原料和300ml甲醇;将4.0g氢氧化钠(4.5倍摩尔量)溶于150ml水中,将制得的氢氧化钠溶液加入到反应瓶中,与反应瓶中的利伐沙班和甲醇混合均匀,在85℃下加热反应5小时,停止反应。用2m盐酸将反应得到的反应液调节至中性,然后用二氯甲烷萃取,萃取的有机相用无水硫酸钠干燥后,旋干有机相,然后将旋干产物真空烘干12h,得到5.2g5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺。
2)取4.5g5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺加入250ml单口瓶,然后加入150ml二氯甲烷和5.23g(4倍摩尔量)二氯亚砜,25℃搅拌5小时停止反应。将得到的反应液分别用饱和碳酸氢钠溶液、饱和nacl溶液洗涤,然后用无水硫酸钠干燥后旋干,柱层析分离得利伐沙班噁唑烷杂质,hplc纯度大于99%。
利伐沙班噁唑烷杂质对照品的核磁共振氢谱图如图1所示,利伐沙班噁唑烷杂质对照品的质谱图如图2所示。由图1和图2可知,本实施例成功制备出式i所示的利伐沙班噁唑烷杂质。
实施例2
利伐沙班噁唑烷杂质对照品的制备方法如下:
1)在500ml反应瓶中加入10.0g利伐沙班固体原料,然后加入300ml甲醇;将2.6g氢氧化钾(2倍摩尔量)溶于300ml水中,将制得的氢氧化钾溶液加入到反应瓶中,与反应瓶中的利伐沙班和甲醇混合均匀,在85℃下加热反应6小时,停止反应。用2m盐酸将反应得到的反应液调节至中性,然后用二氯甲烷萃取,萃取的有机相用无水硫酸钠干燥后,旋干有机相,然后将旋干产物真空烘干12h,得到6.0g5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺。
2)将4.5g5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺加入250ml单口瓶,然后加入150ml三氯甲烷和2.62g(2倍摩尔量)二氯亚砜,室温搅拌8小时停止反应。将得到的反应液分别用饱和碳酸氢钠溶液、饱和nacl溶液洗涤,然后用无水硫酸钠干燥后旋干,柱层析分离得利伐沙班噁唑环杂质,hplc纯度大于99%。
利伐沙班噁唑烷杂质对照品的核磁共振氢谱图如图1所示,利伐沙班噁唑烷杂质对照品的质谱图如图2所示。由图1和图2可知,本实施例成功制备出式i所示的利伐沙班噁唑烷杂质。
实施例3
利伐沙班噁唑烷杂质对照品的制备方法如下:
1)在500ml反应瓶中加入10.0g利伐沙班固体原料,再加入300ml乙醇;将3.3g氢氧化钾(6倍摩尔量)溶于60ml水中,将制得的氢氧化钾溶液加入到反应瓶中,与反应瓶中的利伐沙班和乙醇混合均匀,在85℃下加热反应3小时,停止反应。用2m盐酸将反应得到的反应液调节至中性,然后用二氯甲烷萃取,萃取的有机相用无水硫酸钠干燥后,旋干有机相,然后将旋干产物真空烘干12h,得到5.5g5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺。
2)将4.5g5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺加入250ml单口瓶,然后加入150ml四氯化碳和6.55g(5倍摩尔量)二氯亚砜,在50℃下搅拌5小时停止反应。反应得到的反应液分别用饱和碳酸氢钠溶液、饱和nacl溶液洗涤,然后用无水硫酸钠干燥后旋干,柱层析分离得利伐沙班噁唑环杂质,hplc纯度大于99%。
利伐沙班噁唑烷杂质对照品的核磁共振氢谱图如图1所示,利伐沙班噁唑烷杂质对照品的质谱图如图2所示。由图1和图2可知,本实施例成功制备出式i所示的利伐沙班噁唑烷杂质。
实施例4
利伐沙班噁唑烷杂质对照品的制备方法如下:
1)在500ml反应瓶中加入10.0g利伐沙班固体原料,再加入300ml乙醇;将1.78氢氧化钠(2倍摩尔量)溶于100ml水中,将制得的氢氧化钠溶液加入到反应瓶中,与反应瓶中的利伐沙班和乙醇混合均匀,在50℃下加热反应8h,停止反应。用2m盐酸将反应得到的反应液调节至中性,然后用二氯甲烷萃取,萃取的有机相用无水硫酸钠干燥后,旋干有机相,然后将旋干产物真空烘干12h,得到5.8g5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺。
2)将4.5g5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺加入250ml单口瓶,然后加入150ml四氢呋喃和2.93g(3倍摩尔量)二氯亚砜,在45℃下搅拌6小时停止反应。反应得到的反应液分别用饱和碳酸氢钠溶液、饱和nacl溶液洗涤,然后用无水硫酸钠干燥后旋干,柱层析分离得利伐沙班噁唑环杂质,hplc纯度大于99%。
利伐沙班噁唑烷杂质对照品的核磁共振氢谱图如图1所示,利伐沙班噁唑烷杂质对照品的质谱图如图2所示。由图1和图2可知,本实施例成功制备出式i所示的利伐沙班噁唑烷杂质。
实施例5
利伐沙班噁唑烷杂质对照品的制备方法如下:
1)在500ml反应瓶中依次加入10.0g利伐沙班固体原料和300ml甲醇;将2.75g氢氧化锂(5.0倍摩尔量)溶于150ml水中,将得到的氢氧化锂溶液加入到反应瓶中,与反应瓶中的利伐沙班和甲醇混合均匀,在65℃下加热反应6小时,停止反应。用2m盐酸将反应得到的反应液调节至中性,然后用二氯甲烷萃取,萃取的有机相用无水硫酸钠干燥后,旋干有机相,然后将旋干产物真空烘干12h,得到5.8g5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺。
2)将4.5g5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺加入250ml单口瓶,然后加入150ml二氯甲烷和5.23g(4倍摩尔量)二氯亚砜,25℃搅拌5小时停止反应。反应得到的反应液分别用饱和碳酸氢钠溶液、饱和nacl溶液洗涤,然后用无水硫酸钠干燥后旋干,柱层析分离得利伐沙班噁唑环杂质,hplc纯度大于99%。
利伐沙班噁唑烷杂质对照品的核磁共振氢谱图如图1所示,利伐沙班噁唑烷杂质对照品的质谱图如图2所示。由图1和图2可知,本实施例成功制备出式i所示的利伐沙班噁唑烷杂质。
- 还没有人留言评论。精彩留言会获得点赞!