一种富集甘草提取物中甘草苷及甘草酸类组分及其制备方法与流程
本发明涉及一种中草药加工技术领域,尤其涉及一种富集甘草提取物中甘草苷及甘草酸类组分及其制备方法。
背景技术:
甘草是豆科植物甘草glycyrrhizauralensisfisch.、胀果甘草glycyrrhizainflatabat.或光果甘草glycyrrhizaglabral.的干燥根和根茎。甘草在我国分布广泛,主产于新疆、内蒙古、甘肃、山西、陕西、东北等地。常作佐使药入多种中药复方,味甘,性平,归心、肺、脾、胃经,具有补脾益气,清热解毒,祛痰止咳,缓急止痛,调和诸药的功效。现代药理学研究表明,具有保肝、抗炎、抗菌、抗病毒、镇咳、抗疟、抗氧化、抗癌、免疫调解、降糖和抗血小板凝集等多种活性。甘草化学成分主要为三萜皂苷类和不同类型的黄酮化合物。
三萜皂苷在甘草中含量较高。至今,从3种常用甘草中分离到的皂苷已有几十种,多以葡萄糖糖苷的形式存在,具有良好的水溶性,其苷元多为齐墩果烷型。齐墩果烷型三萜类化合物主要包括甘草酸(甘草甜素)、甘草次酸及其衍生物、乌热酸(18-α甘草次酸)和甘草内酯等。甘草酸(glycyrrhizic)具有多种药理活性。目前,甘草酸被广泛应用于多种肝病的治疗。近年来,甘草酸的保肝机制成为国际研究的焦点,取得了较大的突破和进展。甘草酸经胃肠道的代谢,生成甘草次酸。近年来,发现其代谢产物甘草次酸同样具有多种药理活性。
甘草中黄酮类化合物基本构型有黄酮类、黄酮醇类、二氢黄酮类、查耳酮类、异黄酮类、异黄烷类、异黄烯类、二氢异黄酮类等几类。甘草苷(liquiritin)是具有抗炎活性的黄酮类成分,已经作为消化性溃疡药收入到日本医药品集中,结构上属于二氢黄酮的一种。并且甘草苷有较强的抗抑郁活性,一直是国内外研究的热点。
目前从甘草提取物中纯化甘草苷和甘草酸类物质的方法主要有结晶法、树脂法等。这些纯化方法对酸碱的依赖度较高,如:冰乙酸重结晶等步骤,需要大量应用冰乙酸,且操作繁琐;离子交换树脂的碱液洗脱又需要利用较多氨水,不利于环境保护,污染较大,并且污水处理也会增加较多生产成本。现纯化制备甘草提取中有效成分收率低,不利于甘草资源的高效利用。所以,寻找一种高效、经济、绿色环保且能大规模工业生产的提取甘草物的制备方法迫在眉睫。
有鉴于此,提出本发明。
技术实现要素:
本发明主要提供一种富集甘草提取物中甘草苷及甘草酸类组分及其制备方法,用于解决目前甘草药材提取制备工艺收率低、纯化产品中目标成分含量低、大量有机溶剂使用造成环境污染和生产成本高等问题。
为了实现上述发明目的,本发明一种富集甘草提取物中甘草苷及甘草酸类组分的制备方法,步骤如下:
(1)干燥提取物制备:将甘草药材洗净、干燥、粉碎,加入甘草质量5-40倍的水进行煎煮,滤取药液,药渣再加5-20倍的水进行煎煮,滤取药液,弃去药渣;合并两次药液,进行旋蒸浓缩、干燥,即得甘草提取物干粉;
(2)目标组分富集:用水溶解甘草提取物粉后过滤膜去颗粒物,采用高效液相色谱技术进行纯化制备,收集柱洗脱液,即为富集甘草苷和甘草酸类组分,经浓缩干燥得富集组分粉末。
在本发明的一些具体实施方案中,步骤(1)和(2)过程中所述用水可采用纯化水、蒸馏水或注射用水。
进一步地,在步骤(2)中所用的高效液相色谱技术,其填料基质为硅胶或高分子聚合物;填料配基的碳原子数为1-30的正链烷基中的一种或多种;粒径为8-100μm;孔径为60-300å,比表面积为100-500m2/g。色谱柱内径为20-1600mm,色谱柱床高度为100-1000mm;色谱平衡液中含有一定浓度的有机溶剂和酸,有机溶剂可以为甲醇、乙醇或乙腈,含量可以为5-10%,酸可以为磷酸、甲酸或乙酸,浓度为0.01-0.5%;色谱柱的载样量为固定相重量的0.01-20.0%;色谱的上样和洗脱流速可以为140-350cm/h;色谱纯化洗脱甘草苷及甘草酸类目标组分的有机溶剂浓度梯度可以是等度和线性。
进一步地,在步骤(1)和(2)中所用干燥方式可以为微波干燥、冷冻干燥、真空干燥或喷雾干燥。
本发明还提出一种甘草苷及甘草酸类制备组分,原料主要组成为甘草药材,经上述各实施例中的制备方法提取富集,步骤(2)中获得的目标组分物相比于步骤(1)中获得的提取粉色泽均匀,表面光洁,质地细腻;富集的组分产品加纯化水后迅速溶解,且其溶液澄清透亮,而提取物粉末溶解速度缓慢且溶液浑浊。
基于上述技术方案,采用高效液相制备技术富集甘草提取物中甘草苷及甘草酸组分可显著提高目标物的纯度及有效成分的含量,该方法节约了药材成本,能够提供更有利于工业生产制备条件,并确保工艺的稳定性和安全性,使甘草药材尽可能的发挥其功效。本发明的特点为:采用高效制备液相方法可有效去除脂类、色素、重金属、农残等无效成分,同时避免有机试剂的使用、减少工业生产污染;更重要的是保留了活性成分,能最大化提高甘草药材的药用价值。
具体来说,与现有技术相比,本发明的优点为:
(1)本发明方法整个富集甘草苷和甘草酸组分的制备过程均可以在室温下进行,减少了能源消耗,显著提高目标组分终产品的质量和收率。
(2)本发明方法采用的高效液相色谱分离技术,可以有效的去除与甘草苷及甘草酸类结构类似物等杂质,且能去除农药残留、重金属、内毒素等有毒有害物质,实现了甘草有效组分的高效纯化制备,更适于工业化生产。
(3)本发明方法整个操作过程操作简单,洗脱剂和再生剂均可回收重复利用,节约成本,可用于大规模生产。
附图说明
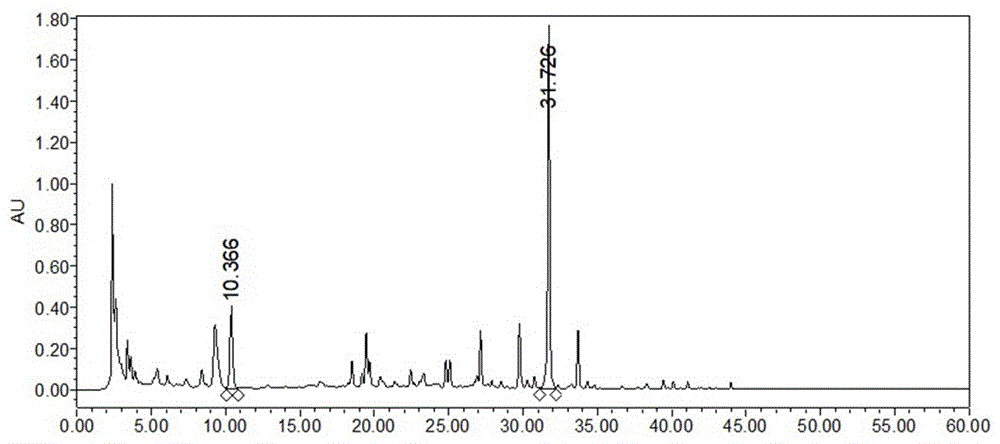
图1为本发明中制备的甘草提取物的hplc分析色谱图;
图2为本发明中富集的甘草苷及甘草酸类组分的hplc分析色谱图。
具体实施方式
下面将结合示意图对本发明富集甘草提取物中甘草苷及甘草酸类组分的制备方法进行更详细的描述,其中表示了本发明的优选实施例,应该理解本领域技术人员可以修改在此描述的本发明,而仍然实现本发明的有利效果。因此,下列描述应当被理解为对于本领域技术人员的广泛知道,而并不作为对本发明的限制。
为了清楚,不描述实际实施例的全部特征。在下列描述中,不详细描述公知的功能和结构,因为它们会使本发明由于不必要的细节而混乱。应当认为在任何实际实施例的开发中,必须做出大量实施细节以实现开发者的特定目标,例如按照有关系统或有关商业的限制,由一个实施例改变为另一个实施例。另外,应当认为这种开发工作可能是复杂和耗费时间的,但是对于本领域技术人员来说仅仅是常规工作。
在本发明中,除另有明确规定和限定,如有术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或隐含指明技术特征的数量。由此,限定有“第一”、“第二”特征可以明示或者隐含包括一个或者多个该特征,在本发明描述中,“至少”的含义是一个或一个以上,除非另有明确具体的限定。
在下列段落中参照附图以举例方式更具体地描述本发明。根据下面说明,本发明的优点和特征将更清楚。需说明的是,附图均采用非常简化的形式且均使用非精准的比例,仅用以方便、明晰地辅助说明本发明实施例的目的。
实施例1
本发明提出一种富集甘草提取物中甘草苷及甘草酸类组分的制备方法,具体步骤如下:
称取预处理好的甘草1kg,加水10l放入煎药机中煮(120℃)1h,纱布过滤,药渣加水10l再次放入煎药机中煮(120℃)0.5h,纱布过滤,合并滤液,旋蒸浓缩后经减压真空干燥得甘草提取物粉末。将甘草提取物粉溶解的样品进行hplc分析,色谱分析结果如图1所示。
将c18反相色谱柱(填料重量300g,粒径60μm)连接到高压液相系统上,设计流速60ml/min(190cm/h),采用5%乙醇平衡缓冲液冲洗色谱柱3bv,将平衡缓冲液溶解的60g甘草提取物粉末(使用0.22μm滤膜去除颗粒性不溶物)通过系统泵流经色谱柱纯化,通过10%乙醇淋洗杂质,60%乙醇洗脱目标物,收集各馏分进行hplc分析,合格馏分合并,即为富集的甘草苷和甘草酸组分。将目标组分旋蒸去乙醇,置于冻干机中直至水分全部去除,收集干燥粉末,即为富集目标组分粉末。称重计算分析甘草苷及甘草酸类组分含量。富集的甘草苷和甘草酸组分的hplc分析结果见图2所示。
实施例2
本发明提出一种一种富集甘草提取物中甘草苷及甘草酸类组分的制备方法,具体步骤如下:
称取预处理好的甘草药材1kg,加水20l放入煎药机中煮(120℃)0.8h,纱布过滤,药渣加水10l再次放入煎药机中煮(120℃)0.4h,纱布过滤,合并滤液,旋蒸浓缩后经喷雾干燥得甘草提取物粉末。
将c18反相色谱柱(填料重量300g,粒径30μm)连接到高压液相系统上,设计流速60ml/min(190cm/h),采用10%乙醇平衡缓冲液冲洗色谱柱2bv,将平衡缓冲液溶解的80g甘草提取物粉末(使用0.22μm滤膜去除颗粒性不溶物)通过系统泵流经色谱柱纯化,通过10%乙醇淋洗杂质,70%乙醇洗脱目标物,收集各馏分进行hplc分析,合格馏分合并,即为富集的甘草苷和甘草酸组分。将目标组分旋蒸去乙醇,低温减压真空干燥直至水分全部去除,收集干燥粉末,即为富集目标组分粉末。称重计算分析甘草苷及甘草酸类组分含量。
实施例3
本发明提出一种富集甘草提取物中甘草苷及甘草酸类组分的制备方法,具体步骤如下:
称取预处理好的甘草药材1kg,加水20l放入煎药机中煮(120℃)1h,纱布过滤,药渣加水10l再次放入煎药机中煮(120℃)0.5h,纱布过滤,合并滤液,旋蒸浓缩后经喷雾干燥得甘草提取物粉末。
将c18反相色谱柱(填料重量300g,粒径10μm)连接到高压液相系统上,设计流速50ml/min(150cm/h),采用5%乙醇平衡缓冲液冲洗色谱柱2bv,将平衡缓冲液溶解的60g甘草提取物粉末(使用0.22μm滤膜去除颗粒性不溶物)通过系统泵流经色谱柱纯化,通过线性梯度增加的方式(10-70%乙醇,40min)洗脱目标物,根据峰型收集各馏分进行hplc分析,合格馏分合并,即为富集的甘草苷和甘草酸组分。将目标组分旋蒸去乙醇,低温减压真空干燥直至水分全部去除,收集干燥粉末,即为富集目标组分粉末。称重计算分析甘草苷及甘草酸类组分含量。
实施例4
本发明提出一种富集甘草提取物中甘草苷及甘草酸类组分的制备方法,具体步骤如下:
称取预处理好的甘草药材1kg,加水10l放入煎药机中煮(120℃)0.8h,纱布过滤,药渣加水10l再次放入煎药机中煮(120℃)0.4h,纱布过滤,合并滤液,旋蒸浓缩后经冷冻干燥得甘草提取物粉末。
将c18反相色谱柱(填料重量300g,粒径30μm)连接到高压液相系统上,设计流速60ml/min(190cm/h),采用5%乙醇平衡缓冲液冲洗色谱柱3bv,将平衡缓冲液溶解的90g甘草提取物粉末(使用0.22μm滤膜去除颗粒性不溶物)通过系统泵流经色谱柱纯化,通过10%乙醇淋洗杂质,60%乙醇洗脱目标物,收集各馏分进行hplc分析,合格馏分合并,即为富集的甘草苷和甘草酸组分。将目标组分旋蒸去乙醇,冷冻干燥直至水分全部去除,收集干燥粉末,即为富集目标组分粉末。称重计算分析甘草苷及甘草酸类组分含量。
上述各实施例中,hplc分析条件如下:
(1)色谱柱填料:unitaryc18(5μm,4.6*150mm);
(2)流动相a:0.05%磷酸水,流动相b:乙腈;
(3)流速:1ml/min;
(4)梯度条件:0-8min19%b相,8-35min从19-50%b相,35-50min从50-100%b相,50-60min100%b相。
(5)柱温:30℃;
(6)检测波长:237nm;
(7)进样体积:10μl。
表1现有技术及各实施例方法得到的甘草苷和甘草酸类组分收率及纯度一览表
本发明还提出一种甘草苷及甘草酸类制备组分,原料主要组成为甘草药材,经上述各实施例中的制备方法提取富集,步骤(2)中获得的目标组分物相比于步骤(1)中获得的提取粉色泽均匀,表面光洁,质地细腻;富集的组分产品加纯化水后迅速溶解,且其溶液澄清透亮,而提取物粉末溶解速度缓慢且溶液浑浊。
参见图1、2,表1及各实施例得到的结果,各步骤中的hplc分析结果表明,提取富集前后样品的分析谱图中目标峰没有显著变化,根据总体积、积分峰面积及重量计算得知,富集后产品中主要成分的含量没有明显下降,甘草苷和甘草酸类组分物质收率达95%以上,纯度可达80%以上,远高于现有技术的收率和纯度。上述实验结果显示,采用本发明中提取富集甘草苷和甘草酸类组分物质的纯化方法制备,获得的富集组分具有高收率和高纯度的优势。
上述仅为本发明的优选实施例而已,并不对本发明起到任何限制作用。任何所属技术领域的技术人员,在不脱离本发明的技术方案的范围内,对本发明揭露的技术方案和技术内容做任何形式的等同替换或修改等变动,均属未脱离本发明的技术方案的内容,仍属于本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!