美洛西林钠杂质A的分离制备方法与流程
本发明涉及一种抗生素类药物化合物杂质的精制方法,具体涉及美洛西林钠杂质a的分离制备方法,属于医药技术领域。
背景技术
美洛西林钠的化学名称为:(2r,5r,6r)-3,3-二甲基-6-[(r)-2[3-(甲黄酰)-2-氧代-1-咪唑烷甲酰氨基]-2-苯乙酰氨基]-7-氧代-4-硫杂-1-氮杂双环[3.2.0]-庚烷-2-甲酸钠盐,其具有下述结构:
美洛西林钠是一种半合成青霉素类抗生素,为白色或类白色粉末或结晶;无臭或稍带特异臭,味苦。在临床上对铜假绿单胞菌、大肠埃希菌、肺炎杆菌、变形杆菌、肠杆菌属、枸橼酸杆菌、沙雷菌属、不动杆菌属以及对青霉素敏感的革兰阳性球菌均有抑制作用。
美洛西林钠属于β-内酰胺青霉素类抗生素,由于β-内酰胺的不稳定性,导致在合成美洛西林钠的过程中不可避免地生成了多种杂质。药物杂质与药品质量、安全性及效能密切相关,因此,杂质控制在药物合成、开发、检测中的重要性受到越来越多的重视。美洛西林钠杂质a的化学结构式如下,目前还没有文献记录和公开报道其合成方法:
在生产美洛西林钠的过程中,需要采用对照品进行杂质a的对照检测。目前国内尚未有研究机构能够提供高效的美洛西林钠杂质a的标准物及制备方法,而进口国外市售产品存在费用高昂、手续繁琐、到货周期长等问题。而且杂质a合成困难,合成后不稳定易降解,且在合成过程中极易产生大量副产物,收率低,导致市售产品纯度不高。
技术实现要素:
本发明的目的在于克服现有技术存在的上述缺陷,提供了优化的美洛西林钠杂质a的制备方法,为美洛西林钠的质量控制和检测提供高纯度对照标准品。
本发明是采用以下的技术方案实现的:
本发明提供美洛西林钠杂质a的分离制备方法,包括以下步骤:
以美洛西林钠为原料,与碱试剂反应制得美洛西林钠杂质a;
所述美洛西林钠的结构如式ⅰ所示:
所述美洛西林钠杂质a的结构如式ⅱ所示:
进一步的,反应结束后,向反应体系中加入酸溶液,调节反应体系的ph值至酸性,再加入无机盐,过滤干燥获得固体。
上述碱试剂为氢氧化钾、氢氧化钠和氨中的一种或几种。
上述氢氧化钾、氢氧化钠的水溶液浓度为0.1-0.5mol/ml,所述氨的水溶液质量百分浓度为25-28%。
上述酸溶液为甲酸、磷酸、醋酸、盐酸及三氟乙酸溶液中的一种或其组合;调节反应体系的ph值为2-5。
为促进反应产物脱水,加入盐作为脱水剂。上述盐可采用常见的无机盐,优选为氢氧化钙、氢氧化钠、氯化钙、氯化钠、氯化钾中的一种或几种。
脱水结束后,洗涤处理产品,洗涤液为水、乙醇、乙醚、丙酮中的一种或几种,洗涤1-2次,获得美洛西林钠杂质a。洗涤次数过多,会造成杂质a损失,影响杂质a产率。
具体的,以式ⅰ所示化合物为原料,加入到碱试剂的水溶液中,在室温下搅拌后,加入酸溶液,调节溶液ph值为2-5,再加入无机盐,过滤洗涤1-2次,干燥后得到美洛西林钠杂质a。本发明中,“室温”指的是环境温度,通常为18℃至35℃,优选22℃至30℃。
本发明还提供了利用液相色谱纯化上述美洛西林钠杂质a的精制方法,包括以下步骤:
(1)供试品溶液的制备:将通过上述步骤制备得到的美洛西林钠杂质a用50%乙腈充分溶解,过滤得到供试品溶液;
(2)液相色谱纯化:将上述供试品溶液经过半制备型液相色谱进行制备纯化,其中色谱条件为:
色谱柱:shim-packgistc18;
流动相:a为0.01%-0.5%体积百分比有机酸水溶液,b为乙腈;流动相a:流动相b=65:35-80:20;
流速:15-20ml/min;
洗脱方式:等度洗脱,流动相a与流动相b保持体积浓度不变,匀速洗脱;
检测波长:210nm以及254nm;
收集色谱峰对应的化合物流份,将所得制备溶液干燥,即得到美洛西林钠杂质a精制品。
优选的色谱条件,步骤(2)中等度洗脱流动相体积比例为流动相a:流动相b=75:25。
进一步的,步骤(2)中流动相a为甲酸水溶液、乙酸水溶液、三氟乙酸水溶液。
与现有技术相比,本发明取得的有益效果是:
(1)本发明的技术方案为一种操作简单的制备美洛西林钠杂质a的精制方法,该技术方案的制备路线短、后处理容易、试剂易得且无污染。
(2)本发明采用半制备型液相色谱,制备分离出的美洛西林钠杂质a,纯度超过95%,远高于市售标准对照品的纯度。同时单次制备时间少于15min,有效缩短了分离时间,且分离度好,工艺简单,有效避免常规方法导致杂质a降解,产品质量稳定,所涉及的溶剂对环境清洁友好。
(3)通过本发明获得的高纯度美洛西林钠杂质a可用于美洛西林钠生产中的杂质定性及定量分析,从而提高美洛西林钠的质量标准,为安全用药提供重要的指导意义。
附图说明
图1是实施例1制备得到的美洛西林钠杂质a利用半制备型液相色谱仪制备所得产物的hplc图。
图2是实施例1制备得到的美洛西林钠杂质a利用半制备型液相色谱仪制备所得产物的hplc图。
图3是实施例2制备得到的美洛西林钠杂质a利用半制备型液相色谱仪制备所得产物的hplc图。
图4是实施例2制备得到的美洛西林钠杂质a利用半制备型液相色谱仪制备所得产物的hplc图。
图5是实施例3制备得到的美洛西林钠杂质a利用半制备型液相色谱仪制备所得产物的hplc图。
图6是实施例4制备得到的美洛西林钠杂质a利用半制备型液相色谱仪制备所得产物的hplc图。
图7是实施例9制备得到的美洛西林钠杂质a利用半制备型液相色谱仪制备所得产物的hplc图。
具体实施方式
下面通过具体实施例对本发明的方法进行说明,但本发明并不局限于此。
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,均采用分析纯试剂,如无特殊说明,均可从商业途径获得。氨水为质量百分浓度为25%-28%,购自顺程化工。所得粗提品的进一步纯化处理由岛津lc-20a半制备高效液相色谱仪进行。
一、美洛西林钠杂质a的分离制备
实施例1
在室温下,向2.0g美洛西林钠中,加入0.1mol/ml的氢氧化钾溶液60ml,搅拌30min后,逐滴滴加0.5mol/ml的盐酸溶液,调节溶液的ph值至3,加入20g氯化钙固体搅拌5min后,过滤,用5ml水淋洗一次,再用5ml乙醇淋洗一次,室温干燥后得到1.1g固体。依据2015年版中国药典测定,用十八烷基硅烷键合硅胶为填充剂;流动相为缓冲液(取磷酸二氢钾4.9g和磷酸氢二钾0.45g,用水溶解稀释至1000ml):乙腈,按体积比80:20等度洗脱,流速1.0ml/min;柱温30℃,检测波长210nm;保留时间11.1min。计算得产率70%。
实施例2
在室温下,向2.0g美洛西林钠中,加入0.1mol/ml的氢氧化钠溶液60ml,搅拌30min后,逐滴滴加磷酸溶液,调节溶液的ph值至4,加入20g氯化钙固体搅拌5min后,过滤,用5ml水淋洗一次,再用5ml乙醇淋洗一次,室温干燥后得到1.1g固体。计算得产率67%。
实施例3
在室温下,向2.0g美洛西林钠中,加入氨水60ml,搅拌30min后,逐滴滴加醋酸溶液,调节溶液的ph值至5,再加入15g氯化钾固体搅拌10min后,过滤,用5ml丙酮淋洗两次,室温干燥后得到1.2g固体,产率52%。
实施例4
在室温下,向2.0g美洛西林钠中,加入0.1mol/ml的氢氧化钾溶液60ml,搅拌40min后,逐滴滴加甲酸溶液,调节溶液的ph值至5,再加入20g氯化钠固体搅拌10min后,过滤,用5ml水淋洗两次,室温干燥后得到1.0g固体,产率55%。
实施例5
在室温下,向2.0g美洛西林钠中,加入氢氧化钾(0.1mol/ml)和氢氧化钠(0.2mol/ml)混合溶液60ml,搅拌40min后,逐滴滴加磷酸溶液,调节溶液的ph值至4,再加入10g氢氧化钠固体搅拌10min后,过滤,用5ml乙醚淋洗两次,室温干燥后得到1.1g固体,产率62%。
实施例6
在室温下,向1.5g美洛西林钠中,加入0.2mol/ml的氢氧化钾溶液40ml,搅拌30min后,逐滴滴加0.5mol/ml的盐酸溶液,调节溶液的ph值至3,再加入10g氯化钠固体搅拌10min后,过滤,用5ml水淋洗一次,再用5ml乙醇淋洗一次,室温干燥后得到0.8g固体,产率65%。
实施例7
在室温下,向1.5g美洛西林钠中,加入0.1mol/ml的氢氧化钾溶液60ml,搅拌40min后,逐滴滴加0.5mol/ml的盐酸溶液,调节溶液的ph值至2,再加入10g氯化钙固体搅拌10min后,过滤,用5ml乙醚淋洗两次,室温干燥后得到0.7g固体,产率51%。
实施例8
在室温下,向1.2g美洛西林钠中,加入0.1mol/ml的氢氧化钠溶液30ml,搅拌20min后,逐滴滴加甲酸溶液,调节溶液的ph值至5,再加入15g氯化钙固体搅拌10min后,过滤,用5ml水淋洗两次,室温干燥后得到0.7g固体,产率58%。
实施例9
在室温下,向1.0g美洛西林钠中,加入0.5mol/ml的氢氧化钠溶液30ml,搅拌20min后,逐滴滴加三氟乙酸溶液,调节溶液的ph值至4,再加入10g氢氧化钙固体搅拌10min后,过滤,用5ml水淋洗两次,室温干燥后得到0.5g固体,产率66%。
实施例10
在室温下,向1.0g美洛西林钠中,加入0.1mol/ml的氢氧化钾溶液60ml,搅拌20min后,逐滴滴加甲酸和醋酸混合溶液,调节溶液的ph值至5,加入20g氯化钙固体搅拌10min后,过滤,用5ml水淋洗一次,再用5ml乙醇淋洗一次,室温干燥后得到0.5g固体,产率54%。
二、美洛西林钠杂质a的精制
通过色谱分离和选择性检测混合物的组分可以对此类混合物进行全面的定性或定量分析。目前,技术人员采用半制备性hplc主要是为了从混合物中富集或纯化目标化合物。但是,半制备型色谱条件与典型的分析型分离所用的条件产生了差异,导致对于复杂样品中目标化合物的富集回收难以采用分析型的色谱条件。实施例9-15利用半制备性液相色谱系统,以及分离纯化条件,对上述实施例中制得美洛西林钠杂质a进行精制。
将上述实施例1-4、9获得的美洛西林钠杂质a精密称定,用50%乙腈溶解,并定量稀释成100mg/ml的溶液,作为供试品溶液。
实施例9
将实施例1的供试品溶液经过半制备型液相色谱进行制备纯化,其中色谱条件为:
色谱柱:shim-packgistc18(20mm×250mm,5μm);
流动相:a为0.01%甲酸水溶液,b为乙腈;流动相a:流动相b=75:25;
流速:15ml/min;
洗脱方式:等度洗脱,流动相a与流动相b保持体积浓度不变,匀速洗脱;
检测波长:210nm以及254nm;
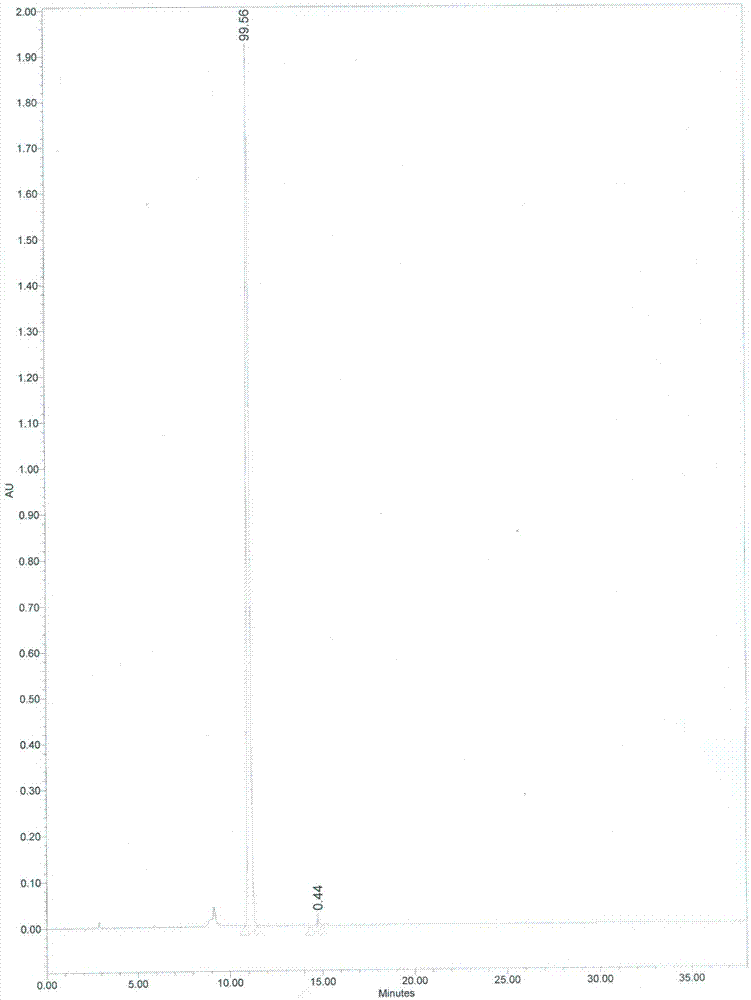
收集色谱峰主峰对应的化合物流份,将所得制备溶液干燥,即得到美洛西林钠杂质a精制品,如图1所示,图1中,美洛西林钠杂质a纯度为99.56%。
实施例10
将实施例1的供试品溶液经过半制备型液相色谱进行制备纯化,其中色谱条件为:
色谱柱:shim-packgistc18(20mm×250mm,5μm);
流动相:a为0.01%乙酸水溶液,b为乙腈;流动相a:流动相b=65:35;
流速:15ml/min;
洗脱方式:等度洗脱,流动相a与流动相b保持体积浓度不变,匀速洗脱;
检测波长:210nm以及254nm;
收集色谱峰主峰对应的化合物流份,将所得制备溶液干燥,即得到美洛西林钠杂质a精制品如图2所示,图2中,美洛西林钠杂质a纯度为88.41%。
实施例11
将实施例2的供试品溶液经过半制备型液相色谱进行制备纯化,其中色谱条件为:
色谱柱:shim-packgistc18(20mm×250mm,5μm);
流动相:a为0.01%甲酸水溶液,b为乙腈;流动相a:流动相b=75:25;
流速:15ml/min;
洗脱方式:等度洗脱,流动相a与流动相b保持体积浓度不变,匀速洗脱;
检测波长:210nm以及254nm;
收集色谱峰主峰对应的化合物流份,将所得制备溶液干燥,即得到美洛西林钠杂质a精制品。如图3所示,图3中,美洛西林钠杂质a纯度为85.39%。
实施例12
将实施例2的供试品溶液经过半制备型液相色谱进行制备纯化,其中色谱条件为:
色谱柱:shim-packgistc18(20mm×250mm,5μm);
流动相:a为0.1%甲酸水溶液,b为乙腈;流动相a:流动相b=75:25;
流速:15ml/min;
洗脱方式:等度洗脱,流动相a与流动相b保持体积浓度不变,匀速洗脱;
检测波长:210nm以及254nm;
收集色谱峰主峰对应的化合物流份,将所得制备溶液干燥,即得到美洛西林钠杂质a精制品。如图4所示,图4中,美洛西林钠杂质a纯度为87.27%。
实施例13
将实施例3的供试品溶液经过半制备型液相色谱进行制备纯化,其中色谱条件为:
色谱柱:shim-packgistc18(20mm×250mm,5μm);
流动相:a为0.5%三氟乙酸水溶液,b为乙腈;流动相a:流动相b=80:20;
流速:18ml/min;
洗脱方式:等度洗脱,流动相a与流动相b保持体积浓度不变,匀速洗脱;
检测波长:210nm以及254nm;
收集色谱峰主峰对应的化合物流份,将所得制备溶液干燥,即得到美洛西林钠杂质a精制品。如图5所示,图5中,美洛西林钠杂质a纯度为63.08%。
实施例14
将实施例4的供试品溶液经过半制备型液相色谱进行制备纯化,其中色谱条件为:
色谱柱:shim-packgistc18(20mm×250mm,5μm);
流动相:a为0.01%三氟乙酸水溶液,b为乙腈;流动相a:流动相b=75:25;
流速:18ml/min;
洗脱方式:等度洗脱,流动相a与流动相b保持体积浓度不变,匀速洗脱;
检测波长:210nm以及254nm;
收集色谱峰主峰对应的化合物流份,将所得制备溶液干燥,即得到美洛西林钠杂质a精制品。如图6所示,图6中,美洛西林钠杂质a纯度为74.41%。
实施例15
将实施例9的供试品溶液经过半制备型液相色谱进行制备纯化,其中色谱条件为:
色谱柱:shim-packgistc18(20mm×250mm,5μm);
流动相:a为0.01%甲酸水溶液,b为乙腈;流动相a:流动相b=75:25;
流速:20ml/min;
洗脱方式:等度洗脱,流动相a与流动相b保持体积浓度不变,匀速洗脱;
检测波长:210nm以及254nm;
收集色谱峰主峰对应的化合物流份,将所得制备溶液干燥,即得到美洛西林钠杂质a精制品。如图7所示,图7中,美洛西林钠杂质a纯度为97.88%。
当然,上述内容仅为本发明的较佳实施例,不能被认为用于限定对本发明的实施例范围。本发明也并不仅限于上述举例,本技术领域的普通技术人员在本发明的实质范围内所做出的均等变化与改进等,均应归属于本发明的专利涵盖范围内。
- 还没有人留言评论。精彩留言会获得点赞!