一种和厚朴酚纳晶的制备方法与流程
本发明属于药物制剂领域,具体的说,涉及一种和厚朴酚纳晶的制备方法。
背景技术:
和厚朴酚(honokiol)是从于木兰科植物厚朴或凹叶厚朴中分离得到的一种联苯二酚类化合物,具有抗炎、抗菌、抗氧化、抗焦虑及抑郁、抗肿瘤、中枢性肌肉松弛、降低胆固醇、抗血小板凝聚等多种生物学活性。转录因子nf-κb可调控炎症反应各阶段的相关分子,目前普遍认为和厚朴酚通过抑制nf-κb的转录活性来发挥抗炎作用。
和厚朴酚水溶性差,生物利用度低,极大地限制了其在临床的应用。现有文献报道在制备和厚朴酚制剂时,大多把和厚朴酚溶在丙酮、甲醇和乙醇等有机溶剂中,存在有机溶剂不易彻底除去的问题。因此,临床上需要一种能增加和厚朴酚水溶性,改善生物利用度,制备工艺简单、安全的制剂。
纳晶也称纳米混悬液,即以表面活性剂或聚合物为稳定剂,将纳米尺度的药物粒子分散在水中形成的稳定胶体分散体系。体系中的药物粒径极小,一般在10-1000nm,这是纳晶与普通制剂最大的区别之一。常用的稳定剂有聚乙二醇、聚乙烯吡咯烷酮、环糊精、纤维素衍生物、表面活性剂如泊洛沙姆和聚山梨醇酯等。根据ostwald-freundlich方程,药物粒径降至纳米级别时,其饱和溶解度显著提高。根据noyes-whitney方程,药物的溶出速率会随着饱和溶解度的增加而增加。因此纳晶能够显著提高药物的饱和溶解度,增加药物的溶出速率,从而提高难溶性药物的口服生物利用度,具有广泛的工业应用前景。目前已成功运用纳晶技术上市的产品有
技术实现要素:
本发明的目的是解决和厚朴酚水溶性差,现有研究中制备工艺复杂、成本高和有机溶剂难除去等问题,提供一种制备过程简单、安全,利于工业化生产、具有良好抗炎活性的和厚朴酚纳晶;进一步来说本发明提供一种和厚朴酚纳晶,粒径小,且pdi小,稳定性高的纳晶。
发明人选用泊洛沙姆407作为稳定剂,并考察了和厚朴酚和泊洛沙姆407在不同质量比时所制备纳晶的粒径及其分布。制备发现,和厚朴酚与泊洛沙姆407的质量比为1:5-1:7时,和厚朴酚纳晶的粒径范围为30-90nm,多分散系数(pdi)较低,稳定性高。当和厚朴酚和泊洛沙姆407的质量比为1:5时效果最佳。
发明人在90℃-100℃加热使和厚朴酚与泊洛沙姆407熔融,并考察了不同加热温度所制备纳晶的粒径及其分布。制备发现,加热温度在90℃-100℃时,和厚朴酚纳晶的粒径范围为30-60nm,pdi较低,稳定性高。当加热温度为90℃时效果最佳。
发明人选用二氧化碳、氮气或惰性气体作为保护气体,并考察保护气体不同时所制备纳晶的静置稳定性。制备发现,保护气体为二氧化碳时,所制备纳晶4℃静置稳定性和粒径分布显著优于保护气体为氮气或惰性气体时。
因此,本发明采用的技术方案是:一种和厚朴酚纳晶,由和厚朴酚和泊洛沙姆407组成,和厚朴酚和泊洛沙姆407的质量比为1:5,加热熔融温度90℃,制备时通入二氧化碳作为保护气体。
本发明的和厚朴酚纳晶可以以液体状态存在,也可以是冻干粉,如果制备冻干粉,需要在制剂中加入冻干保护剂,和厚朴酚与冻干保护剂的质量比为1:4。所述的冻干保护剂为甘露醇。
本发明公开了一种和厚朴酚纳晶的制备方法,包括如下步骤:
a.加热使和厚朴酚与泊洛沙姆407熔融;
b.在上述熔融物中加水,搅拌;
c.高压均质处理,即得和厚朴酚纳晶;
d.和厚朴酚纳晶与冻干保护剂混合,经冷冻干燥,得到和厚朴酚纳晶冻干粉。
所述加热温度优选为90℃-100℃,更优选为90℃。和厚朴酚与泊洛沙姆407的质量比优选为1:5-1:7,更优选为1:5。在气体保护下,在上述熔融物中加水,搅拌。所述保护气体优选为二氧化碳、氮气或惰性气体,更优选为二氧化碳。
本发明所述的和厚朴酚纳晶有如下优点:
(1)本发明使用熔融法处理和厚朴酚,避免了有机溶剂的使用,无需解决有机溶剂残留问题,提高了制剂安全性。
(2)在应用气体保护,特别是使用二氧化碳时,意外发现纳晶的粒径分布特别均匀,稳定性也好。
(3)本发明使用高压均质法,操作简便,易于工业化生产。
(4)本发明使用的泊洛沙姆407为药用辅料,具有良好的安全性。
(5)本发明的和厚朴酚纳晶能明显抑制二甲苯所致小鼠耳肿胀,具有良好的抗炎活性,可应用于炎症等的治疗。
附图说明
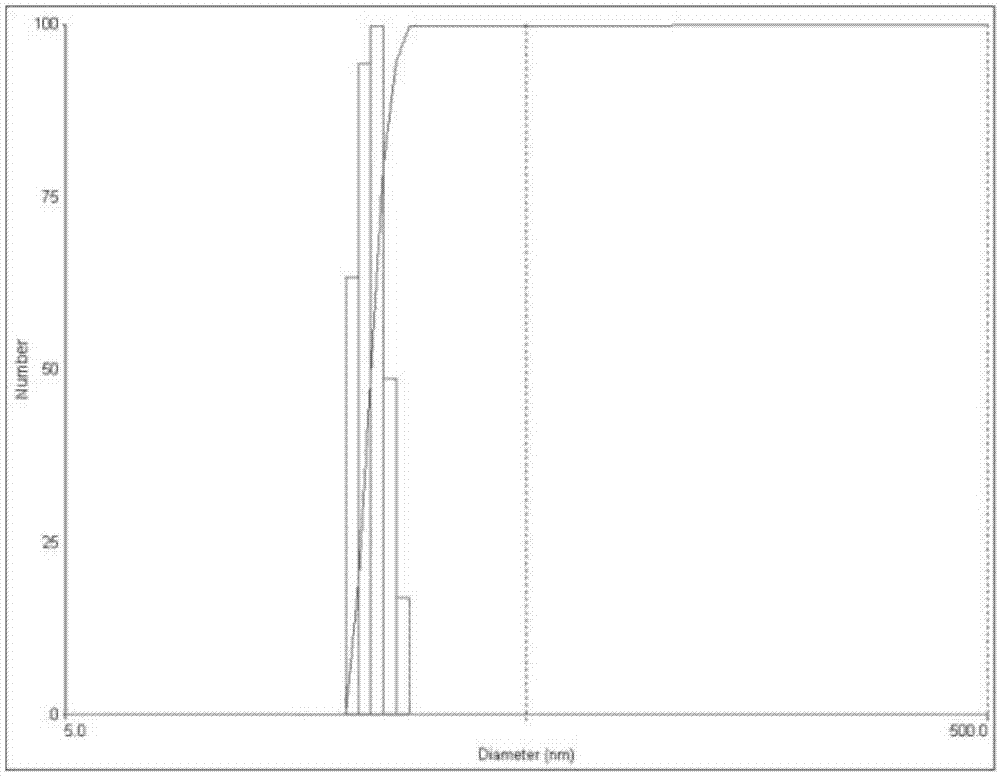
图1是实例1制备的和厚朴酚纳晶的粒径分布图
图2是实例1制备的和厚朴酚纳晶的透射电镜图
具体实施方式
实施例1
称取200mg和厚朴酚,1000mg泊洛沙姆407,90℃加热使熔融。向熔融物中加入80ml水,二氧化碳保护下于90℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。测得平均粒径为30.1nm,pdi为0.12。本实施例的粒径分布图如图1所示。本实施例的透射电镜图如图2所示。和厚朴酚纳晶外形圆整,粒径分布较为均匀。4℃静置30天后测得平均粒径为31.7nm,pdi为0.27。
实施例2
称取200mg和厚朴酚,800mg泊洛沙姆407,90℃加热使熔融。向熔融物中加入80ml水,二氧化碳保护下于90℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。测得平均粒径为102.6nm,pdi为0.38。4℃静置30天后测得平均粒径为117.3nm,pdi为0.46。
实施例3
称取200mg和厚朴酚,1200mg泊洛沙姆407,90℃加热使熔融。向熔融物中加入80ml水,二氧化碳保护下于90℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。测得平均粒径为65.2nm,pdi为0.23。4℃静置30天后测得平均粒径为78.1nm,pdi为0.35。
实施例4
称取200mg和厚朴酚,1400mg泊洛沙姆407,90℃加热使熔融。向熔融物中加入80ml水,二氧化碳保护下于90℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。测得平均粒径为84.6nm,pdi为0.28。4℃静置30天后测得平均粒径为100.2nm,pdi为0.40
实施例5
称取200mg和厚朴酚,1600mg泊洛沙姆407,90℃加热使熔融。向熔融物中加入80ml水,二氧化碳保护下于90℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。测得平均粒径为92.4nm,pdi为0.37。4℃静置30天后测得平均粒径为109.6nm,pdi为0.42。
上述实施例1-5,结果显示:和厚朴酚和泊洛沙姆407在不同质量比时所制备纳晶的粒径及其分布。制备发现,在二氧化碳保护下,和厚朴酚与泊洛沙姆407的质量比为1:5-1:7时,和厚朴酚纳晶的粒径范围为30-90nm,多分散系数(pdi)较低,稳定性高。具体为当和厚朴酚和泊洛沙姆407的质量比为1:5时效果最佳,稳定性最佳,随着和厚朴酚与泊洛沙姆407的质量比增高,粒径变大,但pdi低于0.3,符合要求。但1:4,1:6,1:7,1:8,在4℃静置30天后测得pdi大于0.3。
实施例6
称取200mg和厚朴酚,1000mg泊洛沙姆407,90℃加热使熔融。向熔融物中加入80ml水,二氧化碳保护下于90℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。加入800mg甘露醇,磁力搅拌10min。把3ml样品置于10ml西林瓶中,于-20℃预冻24h,于冷冻干燥机内冻干24h,制成和厚朴酚纳晶冻干粉。冻干粉用pbs复溶后,测得平均粒径为30.4nm,pdi为0.16。4℃静置30天后测得平均粒径为32.0nm,pdi为0.28。
实施例7
称取200mg和厚朴酚,1000mg泊洛沙姆407,95℃加热使熔融。向熔融物中加入80ml水,二氧化碳保护下于95℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。测得平均粒径为42.9nm,pdi为0.24。4℃静置30天后测得平均粒径为50.6nm,pdi为0.37。
实施例8
称取200mg和厚朴酚,1000mg泊洛沙姆407,100℃加热使熔融。向熔融物中加入80ml水,二氧化碳保护下于100℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。测得平均粒径为55.7nm,pdi为0.26。4℃静置30天后测得平均粒径为62.4nm,pdi为0.40。
实施例6-8结果显示:加热温度在90℃-100℃时,和厚朴酚纳晶的粒径范围为30-60nm,pdi较低,稳定性高。当加热温度为90℃时效果最佳,随着温度升高,粒径和pdi都增大。
实施例9
称取200mg和厚朴酚,1000mg泊洛沙姆407,90℃加热使熔融。向熔融物中加入80ml水,氮气保护下于90℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。测得平均粒径为31.5nm,pdi为0.19。4℃静置30天后测得平均粒径为43.7nm,pdi为0.35。
实施例10
称取200mg和厚朴酚,1000mg泊洛沙姆407,90℃加热使熔融。向熔融物中加入80ml水,氩气保护下于90℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。测得平均粒径为37.2nm,pdi为0.22。4℃静置30天后测得平均粒径为45.5nm,pdi为0.34。
实施例11
称取200mg和厚朴酚,1000mg泊洛沙姆407,90℃加热使熔融。向熔融物中加入80ml水,无气体保护下于90℃磁力搅拌1h。将磁力搅拌得到的药物溶液高压均质,条件为400bar循环20次,制得和厚朴酚纳晶溶液。测得平均粒径为32.7nm,pdi为0.15。4℃静置30天后测得平均粒径为49.3nm,pdi为0.35。4℃静置30天后测得平均粒径为60.3nm,pdi为0.46。
实施例9-11,结果显示:比较了二氧化碳、氮气或惰性气体作为保护气体及无气体保护,所制备纳晶的静置稳定性,保护气体为二氧化碳时,所制备纳晶4℃静置稳定性和粒径分布显著优于保护气体为氮气或惰性气体时,无气体保护、氮气或惰性气体的粒径差异不大,但4℃静置30天后测得pdi都高于0.3。
实施例12
选择4~6周龄,体重18~22g,spf级icr小鼠24只,雌、雄各半。将小鼠随机分成四组,每组6只,分别为空白对照组、阳性对照组、和厚朴酚原料药组和和厚朴酚纳晶组。空白对照组灌胃给药游离生理盐水溶液、阳性对照组灌胃给药阿司匹林原料药混悬液(100mg/kg)、和厚朴酚原料药组灌胃给药和厚朴酚原料药混悬液(100mg/kg)、和厚朴酚纳晶组灌胃给药和厚朴酚纳晶溶液(实施例1)(100mg/kg),每日给药一次,连续给药7天。末次给药30min后,将20μl二甲苯均匀涂抹在小鼠右耳前后两面,左耳不涂为正常耳。30min后,将小鼠颈椎脱臼致死,剪下双耳,用直径6mm的打孔器分别在双耳同一部位打下圆耳片,立刻用电子天平称重。
以右耳片与左耳片质量之差计算肿胀度,以(空白对照组平均肿胀度-实验组平均肿胀度)/空白对照组平均肿胀度×100%计算肿胀抑制率。所有数据用ibmspssstatistics22统计软件进行统计分析,各组之间的比较采用单因素方差分析法,p<0.05被认为差异有统计意义,p<0.01被认为差异有高度统计意义。
实验结果参见表1。阳性对照药阿司匹林与和厚朴酚均有抑制二甲苯所致小鼠耳肿胀的作用。其中,阳性对照组肿胀度与和厚朴酚纳晶组肿胀度差异无统计学意义(p>0.05),和厚朴酚原料药组肿胀度与阳性对照组肿胀度、和厚朴酚纳晶组肿胀度差异均有统计意义(p<0.05),表明和厚朴酚纳晶的抗炎效果优于和厚朴酚原料药。
表1不同处理对二甲苯所致小鼠耳肿胀的影响(mean±sd,n=6)
- 还没有人留言评论。精彩留言会获得点赞!