一种合成氟班色林的新方法与流程
本发明属于有机合成技术领域,具体涉及一种合成氟班色林的新方法。
背景技术:
氟班色林(flibanserin),是一种新型的非激素类药物,最早由德国boehringeringelheim公司研制,其中文化学名称为:3-[2-[4-(4-三氟甲基苯基)哌嗪-1-基]乙基]-1h-苯并咪唑-2-酮,结构如下式一所示。
氟班色林是一种解决妇女性功能障碍的药物,主要用于治疗获得性欲障碍(性欲减退症,hsdd)的绝经前妇女。在全球范围内,长期以来受性欲低下困扰的女性占5.4%~13.6%,其中绝经前女性(18~55岁)发病率约为hsdd的10%。氟班色林于2009年11月完成三期临床试验并提出新药申请,2015年8月,该药由fda批准上市,并以
女性性功能障碍(femalesexualdysfunction,简称fsd)是指女性由于性欲低下、性唤起困难、性高潮障碍或性交疼痛使其明显受此困扰的一类疾病。fsd病症中性欲低下(hypoactivesexualdesiredisorder,hsdd)是指非由物质或一般医疗状况所致的持续地或反复地对性生活的欲望以及性幻想不足或完全缺失,且由此给患者带来显著的苦恼或人际交往困难。hsdd是一种常见且难治的精神类泌尿生殖系统疾病,它可发生于各年龄段的女性。kennedy等关于氟班色林治疗重度抑郁症的研究发现,氟班色林虽无抗抑郁作用,但它在提高患者性欲方面显示出了良好的疗效,这促使广大研究者应用氟班色林治疗hsdd。目前,对hsdd的治疗包括三种疗法,如心理治疗、激素治疗和精神药物治疗。精神药物治疗主要是从生理层面上解除其致病因素。在生理因素中,目前研究较为清楚的是中枢神经系统对性反应的兴奋和抑制调节出现异常。在中枢神经系统中,对性反应起兴奋作用的神经介质为多巴胺及去甲肾上激素,而对性反应起抑制作用的神经介质为5-羟基色胺(5-hyroxytryptamine,5-ht)。氟班色林是大脑皮质突触5-ht1a激动剂以及5-ht2a拮抗剂,它与多巴胺d4、5-ht2b及5-ht2c受体均具有中度亲和力。在生理层面上,氟班色林通过与5-ht竞争性结合其在突触后膜上的受体,使得5-ht不能发挥其抑制性欲的效应,进而使得中枢对性反应的抑制性减弱,兴奋性增高,从而达到治疗hsdd的目的。
目前国内外关于氟班色林的制备方法主要有三种:
a、世界专利wo199303016首先报道了氟班色林的合成方法,其通过苯并咪唑酮中间体和哌嗪中间体的取代反应得到,合成路线如式一所示。
b、世界专利wo2010128516公开了一种氟班色林改进制备方法,主要是考虑到哌嗪衍生物难以获得的情况,采用二(2-氯乙基)胺或二乙醇胺与保护的苯并咪唑酮衍生物进行取代缩合得到,合成路线如式二所示。
c、中国专利cn201510391692也公开了一种氟班色林改良制备方法,其采用先制备哌嗪中间体,再缩合成苯并咪唑酮环的方法得到,合成路线如式三所示。
上述三条合成路线制备氟班色林都存在原料难获得,反应步骤多、活性反应点较多,使得副反应难控制,收率降低,工业放大存在困难,导致成本较高等弊端。因此,对该药物制备寻找一条原料易得、工艺简单、收率较高且经济环保的工业化路线很有必要。
技术实现要素:
为了克服现有技术存在的问题,本发明的目的在于提供一种合成氟班色林的新方法。本发明方法分别以三乙醇胺和间氨基三氟甲苯为起始原料,制备了哌嗪中间体;以邻苯二胺和原碳酸四乙酯为原料,制备出了乙氧基苯并咪唑类中间体;然后将制得的哌嗪中间体和苯并咪唑类中间体发生取代反应,以及盐酸脱保护等步骤得到目标产物氟班色林。
本发明提供的合成氟班色林的新方法,所述方法的合成路线如下式四所示:
上述合成氟班色林的新方法,所述方法包括如下步骤:
(1)在装有磁力搅拌子的单口烧瓶中按比例依次加入邻苯二胺、原碳酸四乙酯和乙酸,室温搅拌混合均匀后于70℃条件下继续搅拌3h,然后加入适量无机碱溶液,继续搅拌反应1h,再趁热过滤,用水洗涤有机相后干燥,得到2-乙氧基苯并咪唑;
其中:所述2-乙氧基苯并咪唑结构式如下式五所示:
(2)在装有磁力搅拌子的三口烧瓶中分别投入氯仿和三乙醇胺,混合均匀后,在室温搅拌条件下逐滴加入适量氯化亚砜,然后升温至70℃回流反应3h,再冷却至室温后真空浓缩,洗涤,得到白色产物,即三(2-氯乙基)胺盐酸盐;
其中:所述三(2-氯乙基)胺盐酸盐结构式如下式六所示:
(3)在装有磁力搅拌子的单口烧瓶中按比例依次加入步骤(2)制得的三(2-氯乙基)胺盐酸盐、间氨基三氟甲苯、碳酸钾和适量正丁醇,升温至115~120℃,搅拌回流反应21~25h,反应结束后真空浓缩,残液用二氯甲烷溶解,然后用等体积水洗涤有机相两次,无水硫酸钠干燥后减压除去溶剂,得到黄绿色油状物,再利用柱层析分离提纯,得到n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪;
其中:所述n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪的结构式如下式七所示:
(4)在装有磁力搅拌子的烧瓶中分别按比例加入步骤(3)制得的n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪、步骤(1)制得的2-乙氧基苯并咪唑和适量naoh溶液、异丙醇、水,搅拌混合均匀后于75℃条件下反应2h,冷却至室温后用乙酸乙酯萃取,然后分别用水和饱和食盐水洗涤有机相,干燥后减压浓缩,得到黄棕色油状物;再加入适量异丙醇、浓盐酸,在70℃条件下继续搅拌反应2h,冷却,析出白色晶体,过滤干燥得氟班色林盐酸盐粗品;将粗品溶于乙酸乙酯与水的混合液中,加入氢氧化钠水溶液,调节ph值至碱性,搅拌,分离得到有机层,再用无水硫酸钠干燥,减压除去溶剂,得到白色固体,最后用乙醇重结晶,过滤干燥,即得到本发明所述的目标产物氟班色林。
进一步地,上述技术方案,步骤(1)所述原碳酸四乙酯与邻苯二胺的摩尔比为1.05:1。
进一步地,上述技术方案,步骤(1)所述乙酸与邻苯二胺的摩尔比为(1~1.02):1。
进一步地,上述技术方案,步骤(1)所述无机碱为氢氧化钾或碳酸钾中的任一种。
进一步地,上述技术方案,步骤(2)所述三乙醇胺与氯仿的用量比为1g:16ml。
进一步地,上述技术方案,步骤(2)所述氯仿与氯化亚砜的体积比为10:1。
进一步地,上述技术方案,步骤(3)所述三(2-氯乙基)胺盐酸盐与间氨基三氟甲苯的摩尔比为(1.2~1.3):1。
进一步地,上述技术方案,步骤(3)所述间氨基三氟甲苯与碳酸钾的摩尔比为1:1。
进一步地,上述技术方案,步骤(3)所述间氨基三氟甲苯与正丁醇的用量比为1mmol:(3~5)ml。
进一步地,上述技术方案,步骤(4)中所述n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪与2-乙氧基苯并咪唑的质量比为1:0.22。
与现有技术相比,本发明涉及的合成氟班色林的新方法具有如下有益效果:
(1)本发明方法反应步骤少,副产物少,中间产物和目标产物收率均较高,本发明中间产物2-乙氧基苯并咪唑的收率可达94.2%,目标产物收率可达56.2%,克服了现有技术中该物质的合成步骤繁琐、工艺冗长、活性反应点较多,使得副反应难控制,目标产物收率较低的缺陷;
(2)本发明方法后处理工艺简单,操作方便,仅需通过低温冷却和重结晶的方法即可分离得到目标产物,且分离得到的目标产物纯度高,纯度可达99.6%;
(3)本发明合成方法工艺简单,反应时间短,无需在惰性氛围下进行长时间反应,能耗低,且本发明对反应设备要求低,原料低廉易得,经济环保,因此,本发明合成方法非常适合工业化生产。
附图说明
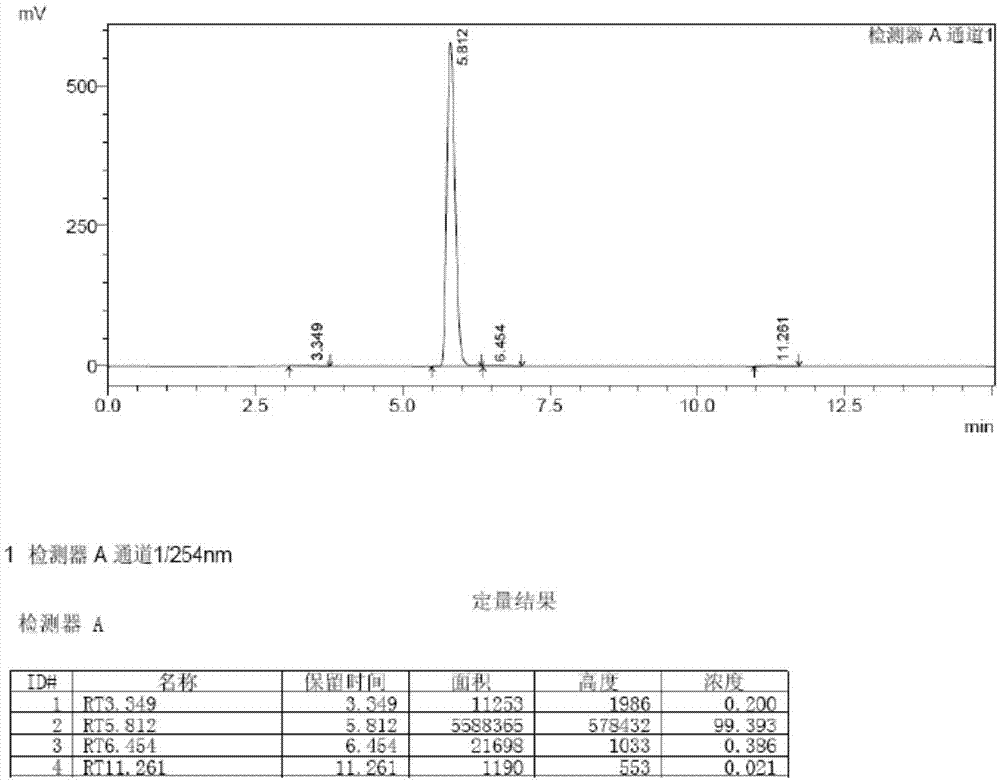
图1为本发明实施例1制备得到的目标产物氟班色林的高效液相色谱(hplc)图;
图2为本发明实施例1制备得到的目标产物氟班色林的esi-ms质谱分析图;
图3为本发明实施例1制备得到的目标产物氟班色林的核磁共振(hnmr)谱图;
图4(a)、(b)分别为本发明实施例1制备得到的中间体n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪的esi-ms质谱分析谱图、高效液相色谱(hplc)图;
图5为本发明实施例1制备得到的中间体2-乙氧基苯并咪唑的高效液相色谱(hplc)图;
图6为本发明实施例1制备得到的中间体2-乙氧基苯并咪唑的esi-ms质谱分析图;
图7为本发明实施例1制备得到的中间体2-乙氧基苯并咪唑的核磁共振(hnmr)谱图。
具体实施方式
下面对本发明的实施案例作详细说明。本实施案例在本发明技术方案的前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施案例。
根据本申请包含的信息,对于本领域技术人员来说可以轻而易举地对本发明的精确描述进行各种改变,而不会偏离所附权利要求的精神和范围。应该理解,本发明的范围不局限于所限定的过程、性质或组分,因为这些实施方案以及其他的描述仅仅是为了示意性说明本发明的特定方面。实际上,本领域或相关领域的技术人员明显能够对本发明实施方式作出的各种改变都涵盖在所附权利要求的范围内。
为了更好地理解本发明而不是限制本发明的范围,在本申请中所用的表示用量、百分比的所有数字、以及其他数值,在所有情况下都应理解为以词语“大约”所修饰。因此,除非特别说明,否则在说明书和所附权利要求书中所列出的数字参数都是近似值,其可能会根据试图获得的理想性质的不同而加以改变。各个数字参数至少应被看作是根据所报告的有效数字和通过常规的四舍五入方法而获得的。
下述各实施例中,中间产物和目标产物纯度的测试条件均为:检测波长:254nm,流动相为25%水和75%乙腈,流速为1ml/min)。
实施例1
本实施例的一种氟班色林的合成新方法,包括如下步骤:
(1)中间体2-乙氧基苯并咪唑的合成
在装有磁力搅拌的100ml单口烧瓶中加入邻苯二胺(16.13g,0.149mol),原碳酸四乙酯(30.02g,0.156mol),乙酸(9.14g,0.152mol)。混合均匀后于70℃搅拌3h。冷却至室温,加入氢氧化钾溶液(5%,w/w,125ml),继续搅拌1h。过滤,用水洗涤后干燥,得到中间体2-乙氧基苯并咪唑21.99g,收率为91.0%,纯度98.8%。
所述中间体2-乙氧基苯并咪唑的hplc检测结果如图5所示,由图5可以看出,保留时间3.3min,纯度98.8%。
由图6和图7可以看出,本实施例上述合成的目标产物的ms和1hnmr测试结果如下:7.38-7.41(m,ar-h,2h),7.14-7.16(m,ar-h,2h),4.59-4.64(q,-ch2,2h),1.44-1.57(m,-ch3,3h)。esi-ms(m/z):163[m+h]+。
(2)中间体三(2-氯乙基)胺盐酸盐的合成
在装有磁力搅拌的250ml三口烧瓶中加入氯仿(200ml)和三乙醇胺(12.5g,83.8mmol),在室温搅拌下逐滴加入氯化亚砜(20ml)。升温至70℃回流反应3小时。冷却至室温后真空浓缩,得到白色固体,用二氯甲烷洗涤三次,得到白色固体即中间体三(2-氯乙基)胺盐酸盐19.87g,收率98.4%。
(3)中间体n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪的合成
在装有磁力搅拌的50ml单口烧瓶中加入三(2-氯乙基)胺盐酸盐(0.3108g,1.3mmol),间氨基三氟甲苯(0.1685g,1.0mmol),碳酸钾(0.139g,1.0mmol),正丁醇(5ml)。加热到115~120℃,搅拌回流反应24h,反应结束后真空浓缩,残液用二氯甲烷溶解。用等体积水洗涤有机相两次,无水硫酸钠干燥后减压除去溶剂,得到黄绿色油状物。再利用柱层析分离提纯(石油醚/乙酸乙酯,8:1),得到n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪0.157g,收率51.3%,纯度98.2%,esi-ms(m/z):293[m+h]+,如图4所示。
(4)目标产物氟班色林的合成
在装有磁力搅拌的50ml烧瓶中加入步骤(3)制得的n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪(0.1g,0.34mmol),步骤(1)制得的2-乙氧基苯并咪唑(0.022g,0.14mmol),45%(w/w)naoh溶液(0.125g),异丙醇0.5ml,水4.4ml,混合均匀后75℃反应2h。冷却至室温,用乙酸乙酯萃取,用水0.4ml和饱和食盐水0.3ml洗涤有机相。干燥后减压浓缩,得到黄棕色油状物。
向上述得到的黄棕色油状物中加入异丙醇3ml,浓盐酸0.68ml,在70℃混合搅拌2h,于4℃冷却,有白色晶体析出,过滤干燥得粗品氟班色林盐酸盐。将粗品溶于乙酸乙酯与水的混合液中,加入氢氧化钠水溶液(5%,w/w),调节ph值至碱性,搅拌30分钟,分离得到有机层,再用无水硫酸钠干燥,减压除去溶剂,得到白色固体。最后用乙醇重结晶,过滤干燥得到目标物氟班色林29.6mg,收率55.8%,纯度99.4%。
将上述实施例1制备得到的目标产物分别采用高效液相色谱(hplc)、esi-ms质谱分析、核磁共振(400mhz,1hnmr)等手段进行表征,测试结果分别参见附图1、图2、图3。
本实施例上述合成的产物的hplc检测结果如图1所示,由图1可以看出,目标产物纯度为99.2%。
由图2和图3可以看出,本实施例上述合成的目标产物的1hnmr和ms测试结果如下:
1hnmr(400mhz,cdcl3),11.02(s,nh,1h),7.31-7.49(t,ar-h,1h),7.29-7.31(d,ar-h,3h),7.15-7.16(d,ar-h,1h),7.03-7.06(m,ar-h,3h),4.28(s,n-ch2,2h),4.00-4.03(d,n-ch2,2h),3.78(s,n-ch2,2h),3.52(s,-ch2,2h),3.24(m,n-ch2,2h),3.14(m,n-ch2,2h).esi-ms(m/z):391[m+1]。
综合上述液相色谱、核磁共振、质谱测试结果,可以确定,本实施例制得的目标化合物为氟班色林,即3-[2-[4-(4-三氟甲基苯基)哌嗪-1-基]乙基]-1h-苯并咪唑-2-酮。
实施例2
本实例的一种氟班色林的合成新方法,包括如下步骤:
(1)中间体2-乙氧基苯并咪唑的合成
在装有磁力搅拌的5000ml单口烧瓶中加入邻苯二胺(0.8kg,7.40mol),原碳酸四乙酯(1.49kg,7.76mol),乙酸(444g,7.40mol)。混合均匀后于70℃搅拌3h。冷却至室温,加入氢氧化钾溶液(5%,6.4l),继续搅拌1h。过滤,用水洗涤后干燥,得到2-乙氧基苯并咪唑1.13kg,收率为94.2%,纯度96.3%。
(2)中间体三(2-氯乙基)胺盐酸盐的合成
在装有磁力搅拌的250ml三口烧瓶中加入氯仿(200ml)和三乙醇胺(12.5g,83.8mmol),在室温搅拌下逐滴加入氯化亚砜(20ml)。升温至70℃回流反应3小时。冷却至室温后真空浓缩,得到白色固体,用二氯甲烷洗涤三次,得到白色固体即中间体三(2-氯乙基)胺盐酸盐19.87g,收率98.4%。
(3)中间体n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪的合成
在装有磁力搅拌的50ml单口烧瓶中加入三(2-氯乙基)胺盐酸盐(2.89g,12.0mmol),间氨基三氟甲苯(1.61g,10.0mmol),碳酸钾(1.38g,10.0mmol),正丁醇(30ml)。加热到115~120℃,搅拌回流反应25h,反应结束后真空浓缩,残液用二氯甲烷溶解。用等体积水洗涤有机相两次,无水硫酸钠干燥后减压除去溶剂,得到黄绿色油状物。柱层析分离提纯(石油醚/乙酸乙酯,8:1),得到n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪0.158g,收率51.7%。纯度98.6%。
(4)目标产物氟班色林的合成
在装有磁力搅拌的50ml烧瓶中加入步骤(3)制得的n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪(0.1g,0.34mmol),步骤(1)制得的2-乙氧基苯并咪唑(0.022g,0.14mmol),45%(w/w)naoh溶液(0.125g),异丙醇0.5ml,水4ml,混合均匀后75℃反应2h。冷却至室温,用乙酸乙酯萃取,用水0.4ml和饱和食盐水0.3ml洗涤有机相。干燥后减压浓缩,得到黄棕色油状物。
向上述得到的黄棕色油状物中加入异丙醇3ml,浓盐酸0.70ml,在70℃混合搅拌2h,于4℃冷却,有白色晶体析出,过滤干燥得粗品氟班色林盐酸盐。将粗品溶于乙酸乙酯与水的混合液中,加入氢氧化钠水溶液(5%,w/w),调节ph值至碱性,搅拌30分钟,分离得到有机层,再用无水硫酸钠干燥,减压除去溶剂,得到白色固体。最后用乙醇重结晶,过滤干燥得到目标物氟班色林29.8mg,收率56.2%,纯度99.4%。
实施例3
本实施例的一种氟班色林的合成新方法,包括如下步骤:
(1)中间体2-乙氧基苯并咪唑的合成
在装有磁力搅拌的5000ml单口烧瓶中加入邻苯二胺(1.60g,14.8mmol),原碳酸四乙酯(3.00g,15.6mmol),乙酸(0.90g,15.0mmol)。混合均匀后于70℃搅拌3h。冷却至室温,加入氢氧化钾溶液(5%,w/w,12.5ml),继续搅拌1h。过滤,用水洗涤后干燥,得到2-乙氧基苯并咪唑2.22kg,收率为92.5%,纯度97.5%。
(2)中间体三(2-氯乙基)胺盐酸盐的合成
在装有磁力搅拌的250ml三口烧瓶中加入氯仿(200ml)和三乙醇胺(12.5g,83.8mmol),在室温搅拌下逐滴加入氯化亚砜(20ml)。升温至70℃回流反应3小时。冷却至室温后真空浓缩,得到白色固体,用二氯甲烷洗涤三次,得到白色固体即中间体三(2-氯乙基)胺盐酸盐19.87g,收率98.4%。
(3)中间体n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪的合成
在装有磁力搅拌的50ml单口烧瓶中加入三(2-氯乙基)胺盐酸盐(2.89g,12.0mmol),间氨基三氟甲苯(1.61g,10.0mmol),碳酸钾(1.38g,10.0mmol),正丁醇30ml。加热到115~120℃,搅拌回流反应23h,反应结束后真空浓缩,残液用二氯甲烷溶解。用等体积水洗涤有机相两次,无水硫酸钠干燥后减压除去溶剂,得到黄绿色油状物。柱层析分离提纯(石油醚/乙酸乙酯,8:1),得到n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪0.156g,收率51.1%。纯度98.1%。
(4)目标产物氟班色林的合成
在装有磁力搅拌的50ml烧瓶中加入步骤(3)制得的n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪(0.1g,0.34mmol),步骤(1)制得的2-乙氧基苯并咪唑(0.022g,0.14mmol),45%(w/w)naoh溶液(0.125g),异丙醇0.5ml,水4ml,混合均匀后75℃反应2h。冷却至室温,用乙酸乙酯萃取,用水0.4ml和饱和食盐水0.3ml洗涤有机相。干燥后减压浓缩,得到黄棕色油状物。
向上述得到的黄棕色油状物中加入异丙醇3ml,浓盐酸0.8ml,在70℃混合搅拌2h,于4℃冷却,有白色晶体析出,过滤干燥得粗品氟班色林盐酸盐。将粗品溶于乙酸乙酯与水的混合液中,加入氢氧化钠水溶液(5%,w/w),调节ph值至碱性,搅拌30分钟,分离得到有机层,再用无水硫酸钠干燥,减压除去溶剂,得到白色固体。最后用乙醇重结晶,过滤干燥得到目标物氟班色林29.4mg,收率55.5%,纯度99.5%。
实施例4
本实例的一种氟班色林的合成新方法,包括如下步骤:
(1)中间体2-乙氧基苯并咪唑的合成
在装有磁力搅拌的100ml单口烧瓶中加入邻苯二胺(16.13g,0.149mol),原碳酸四乙酯(30.02g,0.156mol),乙酸(9.14g,0.152mol)。混合均匀后于70℃搅拌3h。冷却至室温,加入氢氧化钾溶液(5%,w/w,125ml),继续搅拌1h。过滤,用水洗涤后干燥,得到2-乙氧基苯并咪唑21.99g,收率为91.0%,纯度98.8%。
(2)中间体三(2-氯乙基)胺盐酸盐的合成
在装有磁力搅拌的250ml三口烧瓶中加入氯仿(200ml)和三乙醇胺(12.5g,83.8mmol),在室温搅拌下逐滴加入氯化亚砜(20ml)。升温至70℃回流反应3小时。冷却至室温后真空浓缩,得到白色固体,用二氯甲烷洗涤三次,得到白色固体即中间体三(2-氯乙基)胺盐酸盐19.87g,收率98.4%。
(3)中间体n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪的合成
在装有磁力搅拌的50ml单口烧瓶中加入三(2-氯乙基)胺盐酸盐(2.89g,12.0mmol),间氨基三氟甲苯(1.61g,10.0mmol),碳酸钾(1.38g,10.0mmol),正丁醇30ml。加热到115~120℃,搅拌回流反应22h,反应结束后真空浓缩,残液用二氯甲烷溶解。用等体积水洗涤有机相两次,无水硫酸钠干燥后减压除去溶剂,得到黄绿色油状物。柱层析分离提纯(石油醚/乙酸乙酯,8:1),得到n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪0.162g,收率52.8%。纯度98.4%。
(4)目标产物氟班色林的合成
在装有磁力搅拌的50ml烧瓶中加入步骤(3)制得的n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪(0.1g,0.34mmol),步骤(1)制得的2-乙氧基苯并咪唑(0.022g,0.14mmol),45%(w/w)naoh溶液(0.125g),异丙醇0.5ml,水4ml,混合均匀后75℃反应2h。冷却至室温,用乙酸乙酯萃取,用水0.4ml和饱和食盐水0.3ml洗涤有机相。干燥后减压浓缩,得到黄棕色油状物。
向上述得到的黄棕色油状物中加入异丙醇3ml,浓盐酸0.68ml,在70℃混合搅拌2h,于4℃冷却,有白色晶体析出,过滤干燥得粗品氟班色林盐酸盐。将粗品溶于乙酸乙酯与水的混合液中,加入氢氧化钠水溶液(5%,w/w),调节ph值至碱性,搅拌30分钟,分离得到有机层,再用无水硫酸钠干燥,减压除去溶剂,得到白色固体。最后用乙醇重结晶,过滤干燥得到目标物氟班色林29.8mg,收率56.2%,纯度99.6%。
实施例5
本实施例的本实例的一种氟班色林的合成新方法,包括如下步骤:
(1)中间体2-乙氧基苯并咪唑的合成
在装有磁力搅拌的5000ml单口烧瓶中加入邻苯二胺(0.8kg,7.40mol),原碳酸四乙酯(1.49kg,7.76mol),乙酸(444g,7.40mol)。混合均匀后于70℃搅拌3h。冷却至室温,加入氢氧化钾溶液(5%,w/w,6.4l),继续搅拌1h。过滤,用水洗涤后干燥,得到2-乙氧基苯并咪唑1.13kg,收率为94.2%,纯度96.3%。
(2)中间体三(2-氯乙基)胺盐酸盐的合成
在装有磁力搅拌的250ml三口烧瓶中加入氯仿(200ml)和三乙醇胺(12.5g,83.8mmol),在室温搅拌下逐滴加入氯化亚砜(20ml)。升温至70℃回流反应3小时。冷却至室温后真空浓缩,得到白色固体,用二氯甲烷洗涤三次,得到白色固体即中间体三(2-氯乙基)胺盐酸盐19.87g,收率98.4%。
(3)中间体n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪的合成
在装有磁力搅拌的50ml单口烧瓶中加入三(2-氯乙基)胺盐酸盐(2.89g,12.0mmol),间氨基三氟甲苯(1.61g,10.0mmol),碳酸钾(1.38g,10.0mmol),正丁醇30ml。加热到115~120℃,搅拌回流反应21h,反应结束后真空浓缩,残液用二氯甲烷溶解。用等体积水洗涤有机相两次,无水硫酸钠干燥后减压除去溶剂,得到黄绿色油状物。柱层析分离提纯(石油醚/乙酸乙酯,8:1),得到n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪0.154g,收率50.2%,纯度为98.2%。
(4)目标物氟班色林的合成
在装有磁力搅拌的50ml烧瓶中加入步骤(3)制得的n-(3-三氟甲基苯基)-n’-(2-氯乙基)-哌嗪(0.1g,0.34mmol),步骤(1)制得的2-乙氧基苯并咪唑(0.022g,0.14mmol),45%(w/w)naoh溶液(0.125g),异丙醇0.5ml,水4ml,混合均匀后75℃反应2h。冷却至室温,用乙酸乙酯萃取,用水0.4ml和饱和食盐水0.3ml洗涤有机相。干燥后减压浓缩,得到黄棕色油状物。
向上述得到的黄棕色油状物中加入异丙醇3ml,浓盐酸0.7ml,在70℃混合搅拌2h,于4℃冷却,有白色晶体析出,过滤干燥得粗品氟班色林盐酸盐。将粗品溶于乙酸乙酯与水的混合液中,加入氢氧化钠水溶液(5%,w/w),调节ph值至碱性,搅拌30分钟,分离得到有机层,再用无水硫酸钠干燥,减压除去溶剂,得到白色固体。最后用乙醇重结晶,过滤干燥得到目标物氟班色林29.2mg,收率55.1%,纯度99.2%。
上述实施例2~5的中间产物及目标产物的测试方法和测试结果与实施例1基本一致,可以确定,实施例2~5制得的目标化合物为氟班色林,即3-[2-[4-(4-三氟甲基苯基)哌嗪-1-基]乙基]-1h-苯并咪唑-2-酮。
- 还没有人留言评论。精彩留言会获得点赞!