检测乙型肝炎病毒中松弛环状DNA和共价闭合环状DNA突变位点的新方法与流程
本发明属于生物技术领域,涉及一种检测乙型肝炎病毒中松弛环状dna和共价闭合环状dna突变位点的新方法,具体地说,涉及等位基因特异性聚合酶链式反应用于检测乙型肝炎病毒中松弛环状dna和共价闭合环状dna突变位点的新方法。
背景技术:
hbvcccdna不能被有效清除是慢性乙型肝炎难以根治的原因。研究表明hbvrcdna是cccdna形成的必要前体结构,为研究rcdna结构转换时的基因序列变化情况,本发明建立了一种基于pcr技术的简便快速分析方法,用以检测定点突变后的hbvrcdna及结构转换为hbvcccdna后的基因组序列情况,从而确定最终结构转换机制。
通常,要了解特定部位的序列情况,会利用pcr扩增相应片段并进行测序,以确定序列的碱基组成,但这一过程中pcr产物需通过电泳鉴定甚至纯化后才能用于测序分析。
技术实现要素:
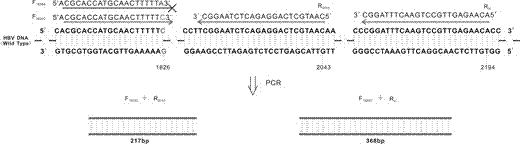
本发明的目的在于提出了一种检测乙型肝炎病毒中松弛环状dna和共价闭合环状dna突变位点的新方法。仅通过pcr后的产物直接电泳即可知相应位点的碱基组成情况。本发明的方法原理是设计上游引物f1826c及f1826a,它们的3’端分别携带定点突变前后相应位点的碱基c(野生型)或a(突变型)。在pcr反应中,由于3’端直接关系dna合成反应的产物延伸,故其在扩增中要求有较严格匹配。结合下游引物r2043及rrc分别扩增同一模板,若模板nt1826位点为野生型,则只有野生型引物对能够扩增出产物,而突变型引物扩增不出产物;反之亦然。同时,为保证设计的单碱基错配引物能够有效促进pcr反应的进行,引物序列应尽可能短且反应条件应小心优化。
其技术方案如下:
一种检测乙型肝炎病毒中松弛环状dna和共价闭合环状dna突变位点的新方法,包括以下步骤:
用来自c1826a之pch9质粒转染hepg2细胞并通过g418筛选得到的hbv复制稳定细胞系中的hbvrcdna及hbvcccdna分别pcr扩增含nt1826的片段,通过t-a克隆,检测菌液模板中nt1826位碱基情况。菌液培养3h,以pbs稀释20倍,其pch9拷贝数/ul即与10pg的pch9质粒拷贝数大致相当,约106copies左右。反应条件选取退火温度63℃,循环数33,行菌液pcr,电泳上样10ul。突变型鉴定采用f1826a/r2043引物对,预期产物大小约200bp;野生型鉴定采用f1826c/rrc引物对,预期产物大小约300bp。
f1826a:5’acgcaccatgcaacttttta3’
f1826c:5’acgcaccatgcaactttttc3’
r2043:5’caatgctcaggagactctaaggc3’
rrc:5’acaagagttgcctgaactttaggc3’
步骤1、设计突变位点鉴别引物两对,突变位点位于正向引物3’末端,如该方法测试引物对f1826a和f1826c,两对引物(突变型与野生型)扩增产物应有明显的长度大小区分,可用电泳鉴别,如该方法中的200bp产物和300bp产物。引物用量,10pmol/ul正反向引物各1ul。
步骤2、扩增模板dna用量以10pg(即约106copies)为最佳,故需将模板配置为10pg/ul(106copies基因数/ul相当的质量或体积)用以pcr扩增,模板用量1ul。突变位点引物与野生型位点引物均需分别扩增检测标本dna。
步骤3、pcr最佳反应条件:选取退火温度63℃,循环数33。即反应条件设置为95℃5min;95℃20sec,63℃20sec,72℃20sec×33;72℃3min;4℃forever。
步骤4、针对上述条件,反应体系如下:
步骤5、将所得产物10ul上样,进行琼脂糖凝胶电泳,若检测标本dna相应位点为野生型,则只有野生型引物对能够扩增出产物,而突变型引物扩增不出产物,反之亦然,以此鉴别检测标本dna中相应位点突变状况。
进一步,为保证设计的单碱基错配引物能够有效促进pcr反应的进行,引物序列应尽可能短且反应条件应小心优化。以上给出的反应条件和参数标准在必要时还要做细微调整。
本发明的有益效果为:
本发明是一种简便、易行、稳定、可靠的检测系统。在野生型模板、突变型模板及实际检测样本中分别应用,优化检测条件后,以模板dna用量为约106copies左右,退火温度为63℃时呈现出较好的区分度。然而,这一检测方法亦非尽善尽美,在区分度方面,似由于g:c间较a:t具有更大的分子结合力,故而对野生型区分度优于突变型。对于大规模样本的定点突变位点分析,由于检测情况简单,故应用本发明研究中所设计的方法,快速准确,因此不失为一种良好的检测系统。
附图说明
图1是pcr鉴定点突变原理示意图及引物序列;
图2是10ng野生型模板pcr扩增结果;1:f1826a/r2043扩增产物;2:f1826a/rrc扩增产物;3:f1826c/r2043扩增产物;4:f1826c/rrc扩增产物;5:阴性对照;m:dl2000marker
图3是10pg野生型模板退火温度优化;1、7、13、5、11、17:突变型引物对f1826a/r2043扩增产物;2、8、14:突变型引物对f1826a/rrc扩增产物;3、9、15、6、12、18:野生型引物对f1826c/r2043扩增产物;4、10、16:野生型引物对f1826c/rrc扩增产物;m:dl2000marker;
图4是c1826a突变型质粒模板反应条件优化;
图5是ta克隆菌液模板鉴定结果;a1-a6:ta克隆菌液20倍稀释模板;m:dl2000marker。
具体实施方式
下面结合附图和具体实施方式对本发明的技术方案作进一步详细地说明。
引物序列及反应原理如图1所示。
首先,本发明利用野生型pch9质粒(含hbv1.1倍体)作为模板,野生型鉴别引物对f1826c/r2043,f1826c/rrc及突变型鉴别引物对f1826a/r2043,f1826a/rrc扩增后电泳检测分析。为得到最佳分辨效果,本发明主要针对模板用量及退火温度进行优化,采用10pg、10ng用量,退火温度采用68℃、63℃、58℃,pcr反应体系:2xgotaqgreenmastermix(promega)10ul,10pmol正反向引物各1ul,模板1ul,ddh2o补足20ul。10ng模板,反应条件采用降落pcr,具体如下:95℃,5min;95℃20sec,65℃(-1℃/cycle)20sec,72℃20sec×10;95℃20sec,55℃20sec,72℃20sec×25;72℃5min;4℃forever。电泳结果如图2示,突变型引物对f1826a/r2043、f1826a/rrc及野生型引物对f1826c/r2043、f1826c/rrc均能扩增出目的大小产物,但突变型及野生型未能作出有效区分。10pg模板,反应条件:95℃5min;95℃20sec,68℃or63℃or58℃20sec,72℃20sec×35;72℃3min;4℃forever。电泳结果如图3示,在退火温度63℃及58℃时,鉴定突变型引物对和鉴定野生型引物对呈现良好区分。
其次,本发明利用突变型c1826a之pch9质粒模板,亦采用10pg(10pg/ul)、10ng(10ng/ul)用量,退火温度采用72℃、68℃、63℃,其它条件与野生型模板扩增相同。应用野生型鉴别引物对f1826c/r2043,f1826c/rrc及突变型鉴别引物对f1826a/r2043,f1826a/rrc扩增后电泳检测分析。如图4示,当退火温度为63℃,模板用量10pg时,鉴定突变型和鉴定野生型引物对对突变质粒做出良好区分。
最终,本发明用来自c1826a之pch9质粒转染hepg2细胞并通过g418筛选得到的hbv复制稳定细胞系中的hbvrcdna及hbvcccdna分别pcr扩增含nt1826的片段,通过t-a克隆,检测菌液模板中nt1826位碱基情况。菌液培养3h,以pbs稀释20倍,其pch9拷贝数/ul即与10pg的pch9质粒拷贝数大致相当,约106copies左右。反应条件选取退火温度63℃,循环数33,行菌液pcr,电泳上样10ul。突变型鉴定采用f1826a/r2043引物对,预期产物大小约200bp;野生型鉴定采用f1826c/rrc引物对,预期产物大小约300bp。随机挑选6个ta克隆菌液样本先行pcr鉴定,电泳上样10ul,结果如图5示。
对ta克隆菌液模板,本发明同时随机选取部分hbvrcdna及hbvcccdna样本共计25例进行测序分析,将测序结果与电泳结果进行比对分析,同一样本采用鉴定野生型引物对及鉴定突变型引物对pcr扩增后电泳分析结果与测序结果完全一致,突变型与野生型区分度良好。
因此,这种基于pcr检测突变位点的方法是一种简便、易行、稳定、可靠的检测系统。在野生型模板、突变型模板及实际检测样本中分别应用,优化检测条件后,以模板dna用量为约106copies左右,退火温度为63℃时呈现出较好的区分度。然而,这一检测方法亦非尽善尽美,在区分度方面,似由于g:c间较a:t具有更大的分子结合力,故而对野生型区分度优于突变型。对于大规模样本的定点突变位点分析,由于检测情况简单,故应用本发明研究中所设计的方法,快速准确,因此不失为一种良好的检测系统。
以上所述,仅为本发明较佳的具体实施方式,本发明的保护范围不限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可显而易见地得到的技术方案的简单变化或等效替换均落入本发明的保护范围内。
序列表
<110>川北医学院
<120>检测乙型肝炎病毒中松弛环状dna和共价闭合环状dna突变位点的新方法
<160>4
<170>siposequencelisting1.0
<210>1
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>1
acgcaccatgcaacttttta20
<210>2
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>2
acgcaccatgcaactttttc20
<210>3
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>3
caatgctcaggagactctaaggc23
<210>4
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>4
acaagagttgcctgaactttaggc24
- 还没有人留言评论。精彩留言会获得点赞!