一种多取代吡啶环类化合物其制备方法及应用与流程
本发明属于药物合成的技术领域,具体涉及一种具有抗肿瘤活性的,具体为一种多取代吡啶环类化合物其制备方法及应用。
背景技术:
含氮杂环化合物由于具有广泛的生物活性而倍受重视,现已开发出许多含氮杂环类的杀菌剂、除草剂、杀虫剂及医用药物,此类化合物已成为农用化学和医药的研究热点,具有广阔的应用前景。例如血红素、叶绿素、dna及rna中的碱基、酶和辅酶中催化生化反应的活性部位及中草药的有效成分生物碱等,都是含氮杂环化合物;为数不多的维生素、抗生素以及一些植物色素和植物染料都含有杂环化。目前合成的杂环化合物涉及医药、农药、染料、生物模拟材料、分子器件、储能材料等,尤其在现代药物中,杂环化合物占有有相当大的比重,与人们的生活息息相关。药物治疗方面成熟的杂环化合物有抗高血压系统药物卡托普利,雷米普利、抗心绞痛药哌嗪类衍生物曲美他嗪、调血脂药氟伐他汀、环吡咯酮类镇静催眠药佐匹克隆、咪唑吡啶类催眠药唑吡坦等。
吡啶化合物是具有良好生物活性的含氮杂环化合物,广泛应用于医药和农药的合成研究。吡啶主要用于合成医药、农药和橡胶助剂,吡啶类农药被称为第四代农药,具有高效、低毒的特点,与人和生物具有良好的环境相容性,符合农药的发展要求和趋势,含吡啶环的化合物逐渐成为农药创制的主要方向之一,非常具有发展前景。例如在医药行业用于抗癌药物伊马替尼,抗结核病的异烟肼等。在橡胶助剂行业用于合成秋兰姆类超级硫化促进剂四硫化双五亚甲基秋兰姆,二硫代氨基甲酸盐类超级促进剂五亚甲基二硫代氨基甲酸哌啶盐等。另外吡啶还可以合成多种新型精细化工中间体,许多产品都属于新开发的小吨位、高附加值的医药、农药和助剂的中间体,如2-甲基吡啶、3-氨甲基吡啶、4-羟基吡啶等。还例如四氢吡啶并[3,4-d]嘧啶化合物可以作为5ht2a受体拮抗剂、细胞外信号调节蛋白激酶抑制剂、哺乳动物p2x7调节剂,以及具有抗乳腺癌细胞mda-mb-231增殖活性和抑制肝癌细胞hepg2的增殖。
我们公司和淄博职业学院合作开发了一种的具有抗肿瘤活性的多吡啶环化合物,并进行了相应的生物活性检测。
技术实现要素:
本发明解决的技术问题是提供了一种多取代吡啶环类化合物其制备方法及应用,该多取代吡啶环类化合物合成方法简单、原料价格低廉,结构新颖,具有抗肿瘤活性。
本发明为解决上述技术问题采用如下技术方案:
一种具有抗肿瘤活性的多取代吡啶环类化合物,其特征在于,其分子结构为:
该化合物的核磁,质谱及元素分析性能参数:1hnmr(400mhz,cdcl3):δ9.37(d,j=12.0hz,1h),8.89-8.81(m,4h),8.53-8.49(m,2h),7.47-7.44(m,4h),7.39-7.31(m,3h),6.14(d,j=4.0hz,1h),5.92(d,j=8.0hz,1h),4.11(s,1h),2.60-2.55(m,2h),2.15(s,1h).hrms(esi):409.1735[m+h]+.anal.calcdforc24h20n6o:c,70.57;h,4.94;n,20.58.found:c,70.25;h,4.89;n,20.71。
本发明的另一目的在于提供一种具有抗肿瘤活性的多取代吡啶环类化合物的制备方法,其特征在于,具体步骤为:
(1)、4-羟基吡啶与乙酸叔丁酯在锰催化剂作用下,发生烯基化反应得到e-3-(吡啶-4-基)丙烯酸叔丁酯;
(2)、e-3-(吡啶-4-基)丙烯酸叔丁酯在氨气氛围下发生迈克尔加成反应得到r-3-氨基-3-(吡啶-4-基)-丙酸叔丁酯;
(3)、r-3-氨基-3-(吡啶-4-基)-丙酸叔丁酯与4-吡啶甲醛发生胺醛缩合反应得到r-3-(4-吡啶甲胺基)-3-(吡啶-4-基)-丙酸叔丁酯;
(4)、r-3-(4-吡啶甲胺基)-3-(吡啶-4-基)-丙酸叔丁酯在酸性条件下水解后再与氨气在铜催化下反应得到
(5)、尿素与1-(吡啶-3-基)丙-2-炔-1-酮在乙醇溶剂中,加热回流制备得到(1-(1-(吡啶基-3-基)-2-亚苄基))尿素;
(6)、(1-(1-(吡啶基-3-基)-2-亚苄基))尿素在多聚磷酸中加热,制得4-(吡啶基-3-基)嘧啶-2(h)-酮;
(7)、4-(吡啶基-3-基)嘧啶-2(h)-酮在甲苯溶剂中,与pocl3反应,制得2-氯-4-(吡啶-3-基)吡啶;
(8)、
进一步优选,步骤(1)的具体过程为:在室温和惰性气体保护下把4-羟基吡啶和乙酸叔丁酯加入叔丁醇中,再加入五羰基溴化锰和叔丁醇钾,缓慢升温至60~90℃反应至原料消失;降温至内温0~5℃,滴加冰乙酸调节反应液ph为中性,过滤反应液,滤液浓缩后得到e-3-(吡啶-4-基)丙烯酸叔丁酯;作为优选4-羟基吡啶与乙酸叔丁酯和叔丁醇钾的投料量摩尔比为1:1.2:1。
进一步优选,步骤(2)的具体过程为:在高压釜中把e-3-(吡啶-4-基)丙烯酸叔丁酯和羟胺加入到水和n,n-二甲基甲酰胺的混合液中,再加入氯化汞,用氮气置换高压釜中的气体两次,再通入氨气,使高压釜中的压力达到压强0.1~0.3mpa,缓慢升温至130℃,反应至原料反应完全,过滤反应液,用盐酸溶液调节反应液ph为4~5,有大量固体析出,抽滤反应液,滤饼经冷甲醇洗涤两次,滤饼放入thf中,再用三乙胺调节反应液ph为8~9,过滤反应液,滤液浓缩后得到r-3-氨基-3-(吡啶-4-基)-丙酸叔丁酯;作为优选e-3-(吡啶-4-基)丙烯酸叔丁酯与羟胺的投料量摩尔比为1:2;e-3-(吡啶-4-基)丙烯酸叔丁酯与氯化汞的投料量质量比为5:1。
进一步优选,步骤(3)的具体过程为:把r-3-氨基-3-(吡啶-4-基)-丙酸叔丁酯和4-吡啶甲醛加入甲苯中,再加入哌啶,接上回流冷凝管和分水器,将混合物搅拌加热至100℃,前期需要溶剂除水,然后反应至原料反应完全后降至室温,分液,将下层液体用少量甲苯萃取多次,与上层液体合并,浓缩反应液得到r-3-(4-吡啶甲胺基)-3-(吡啶-4-基)-丙酸叔丁酯。
进一步优选,步骤(4)的具体过程为:把r-3-(4-吡啶甲胺基)-3-(吡啶-4-基)-丙酸叔丁酯加入到有机酸中,加热到60℃反应至2~4h,然后整除有机酸,转移至高压反应釜中,加入四氢呋喃和氯化亚铜,通入氨气,使釜内压力达到0.1mpa,加热至回流,反应结束后先在减压条件下,蒸去绝大部分的溶剂四氢呋喃,经硅胶柱层析分离得纯净的
进一步优选,步骤(5)的具体过程为:尿素溶于无水乙醇中,加热50℃,然后缓慢滴加1-(吡啶-3-基)丙-2-炔-1-酮,滴完后,加热至轻微回流,然后反应至有大量的白色固体析出,tlc检测原料,原料消失完全,白色固体经过滤,烘干得类白色固体(1-(1-(吡啶基-3-基)-2-亚苄基))尿素。
进一步优选,步骤(6)的具体过程为:在多聚磷酸中,然后加入(1-(1-(吡啶基-3-基)-2-亚苄基))尿素在室温搅拌10~30分钟后加热至110℃,反应结束后,缓慢加入水和活性炭,搅拌均匀后过滤,滤液放冰箱过夜,然后通过真空过滤收集沉淀的固体,用水将固体进行重结晶,烘干得到固体4-(吡啶基-3-基)嘧啶-2(h)-酮。
进一步优选,步骤(7)的具体过程为:在甲苯溶剂中,然后加入4-(吡啶基-3-基)嘧啶-2(h)-酮,在室温搅拌10~30分钟后缓慢滴加三氯氧磷,加热至轻微回流,搅拌反应至原料完全消失后,反应液降温至室温,然后把反应液倒入大量冰水中淬灭反应,加入有机溶剂二氯甲烷,萃取后合并有机相,然后浓缩,通过柱层析纯净的2-氯-4-(吡啶-3-基)吡啶。
进一步优选,步骤(8)的具体过程为:
本发明新方法合成的多取代吡啶环类化合物,具有抗肿瘤活性,并进行了相应的生物活性检测。
本发明还提供了多取代吡啶环类化合物在制备治疗癌症药物中的应用,所述的癌症涉及非小细胞肺癌。
本发明的有益效果为:
本发明的化合物具有抗肿瘤活性,该化合物分子能够有效的进入肺癌细胞的靶点蛋白中,被靶点蛋白完全包括,并与周围的氨基酸发生相应的作用力,进而与该蛋白形成非常显著的作用效果,能够抑制该蛋白的活性,从而抑制肿瘤细胞的生长,具有成药潜质。
本发明的具有抗肿瘤活性的多取代吡啶环类化合物,合成方法简单、原料价格低廉,结构新颖。
附图说明
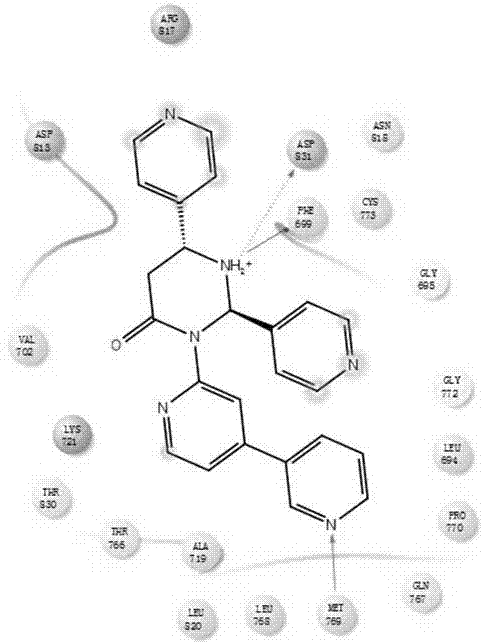
图1为本发明实施例12的药物分子和肿瘤靶点对接图。
具体实施方式
以下通过实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容实现的技术均属于本发明的范围。
实施例1
在反应瓶中,氮气保护下,开动冷却,内温20~25℃,把4-羟基吡啶11g和乙酸叔丁酯14g滴加到叔丁醇100ml中,滴加完后加入五羰基溴化锰1g和叔丁醇钾11g;保持室温搅拌10min,缓慢升温至80℃,继续反应1h,tlc显示原料消失,降温至内温0~5℃,滴加冰乙酸调节反应液ph为中性,过滤反应液,滤液浓缩后得到e-3-(吡啶-4-基)丙烯酸叔丁酯16g;1hnmr(400mhz,dmso-d6):δ8.71(s,2h),7.68-7.64(m,2h),7.37(d,j=12.0hz,1h),5.29(d,j=8.0hz,1h),1.25(s,9h);13cnmr(101mhz,dmso-d6):165.2,157.9,144.3,141.6,127.4,123.8,96.5,28.3。
实施例2
在反应瓶中,氮气保护下,开动冷却,内温20~25℃,把4-羟基吡啶11g和乙酸叔丁酯14g滴加到叔丁醇100ml中,滴加完后加入五羰基溴化锰1g和叔丁醇钾11g;保持室温搅拌10min,缓慢升温至60℃,继续反应1h,tlc显示原料消失,降温至内温0~5℃,滴加冰乙酸调节反应液ph为中性,过滤反应液,滤液浓缩后得到e-3-(吡啶-4-基)丙烯酸叔丁酯7g;1hnmr(400mhz,dmso-d6):δ8.71(s,2h),7.68-7.64(m,2h),7.37(d,j=12.0hz,1h),5.29(d,j=8.0hz,1h),1.25(s,9h);13cnmr(101mhz,dmso-d6):165.2,157.9,144.3,141.6,127.4,123.8,96.5,28.3。
实施例3
在反应瓶中,氮气保护下,开动冷却,内温20~25℃,把4-羟基吡啶11g和乙酸叔丁酯14g滴加到叔丁醇100ml中,滴加完后加入五羰基溴化锰1g和叔丁醇钾11g;保持室温搅拌10min,缓慢升温至90℃,继续反应1h,tlc显示原料消失,降温至内温0~5℃,滴加冰乙酸调节反应液ph为中性,过滤反应液,滤液浓缩后得到e-3-(吡啶-4-基)丙烯酸叔丁酯14g;1hnmr(400mhz,dmso-d6):δ8.71(s,2h),7.68-7.64(m,2h),7.37(d,j=12.0hz,1h),5.29(d,j=8.0hz,1h),1.25(s,9h);13cnmr(101mhz,dmso-d6):165.2,157.9,144.3,141.6,127.4,123.8,96.5,28.3。
实施例4
在高压釜中把e-3-(吡啶-4-基)丙烯酸叔丁酯20g和羟胺6.5g加入到水100g和n,n-二甲基甲酰胺100g的混合液中,再加入氯化汞4g,用氮气置换高压釜中的气体两次,再通入氨气,使高压釜中的压力达到0.2mpa,缓慢升温至130℃,反应至tlc监控原料反应完全,过滤反应液,用盐酸溶液调节反应液ph为4~5,有大量固体析出,抽滤反应液,滤饼经冷甲醇洗涤两次,滤饼放入thf中,再用三乙胺调节反应液ph为8~9,过滤反应液,滤液浓缩后得到r-3-氨基-3-(吡啶-4-基)-丙酸叔丁酯20g,e.e值为99%,hrms(esi):223.1492[m+h]+。
实施例5
在反应瓶中,把r-3-氨基-3-(吡啶-4-基)-丙酸叔丁酯22g和4-吡啶甲醛12g加入甲苯200ml中,再加入哌啶13g,接上回流冷凝管和分水器,将混合物搅拌加热至100℃,通过分水器除去试剂包含的及反应中产生的水,然后继续反应1h,tlc监控原料反应完全后降至室温,分液,将下层液体用少量甲苯萃取多次,与上层液体合并,浓缩反应液得到r-3-(4-吡啶甲胺基)-3-(吡啶-4-基)-丙酸叔丁酯25g,e.e值为99.5%,1hnmr(400mhz,dmso-d6):δ8.93-8.86(m,2h),8.71-8.65(m,2h),8.12(t,j1=4.0hz,j2=12.0hz,1h),7.93(d,j=4.0hz,1h),7.32(d,j=4.0hz,2h),7.07(s,1h),4.15(d,j=8.0hz,1h),2.93-2.88(m,2h).1.31(s,9h)。
实施例6
把r-3-(4-吡啶甲胺基)-3-(吡啶-4-基)-丙酸叔丁酯31g加入到无水甲酸150ml中,加热到60℃反应2h,然后真空整除甲酸,把浓缩物转移至高压反应釜中,加入四氢呋喃200ml和氯化亚铜5g,通入氨气使反应釜内压力达到0.1mpa,加热至60℃,反应结束后,过滤反应液,滤液经减压条件下,蒸去绝大部分的溶剂四氢呋喃,经硅胶柱层析分离得纯净的
实施例7
把r-3-(4-吡啶甲胺基)-3-(吡啶-4-基)-丙酸叔丁酯31g加入到无水乙酸150ml中,加热到60℃反应2h,然后真空整除乙酸,把浓缩物转移至高压反应釜中,加入四氢呋喃200ml和氯化亚铜5g,通入氨气使反应釜内压力达到0.1mpa,加热至60℃,反应结束后,过滤反应液,滤液经减压条件下,蒸去绝大部分的溶剂四氢呋喃,经硅胶柱层析分离得纯净的
实施例8
在1000ml的圆底烧瓶中,把尿素60g溶于无水乙醇300ml中,加热50℃,然后缓慢滴加1-(吡啶-3-基)丙-2-炔-1-酮160g滴完后,加热至轻微回流,然后反应3小时,有大量的白色固体析出,白色固体经过滤,烘干得类白色固体(1-(1-(吡啶基-3-基)-2-亚苄基))尿素160g;1hnmr(400mhz,cdcl3):9.23(d,j=8.0hz,1h),8.83(d,j=24.0hz,1h),8.39(d,j=24.0hz,1h),7.62(t,j1=8.0hz,j2=8.0hz,1h),6.04(s,1h),1.83(s,1h)
实施例9
在3000ml的圆底烧瓶中,加入多聚磷酸800g,然后加入(1-(1-(吡啶基-3-基)-2-亚苄基))尿素180g,搅拌20分钟,加热至110℃,搅拌1小时,缓慢加入水1500ml和活性炭30g,搅拌1分钟,过滤,滤液放冰箱过夜,然后通过真空过滤收集沉淀的固体,用水将固体进行重结晶,烘干得到固体4-(吡啶基-3-基)嘧啶-2(h)-酮161g;1hnmr(400mhz,cdcl3):9.16(s,1h),8.79(d,j=12.0hz,1h),8.42(d,j=12.0hz,1h),7.67(t,j1=8.0hz,j2=8.0hz,1h),7.31(d,j=24.0hz,1h),4.57(d,j=16.0hz,1h)。
实施例10
在3000ml的圆底烧瓶中,加入甲苯800ml,然后加入4-(吡啶基-3-基)嘧啶-2(h)-酮170g室温搅拌10分钟,缓慢滴加三氯氧磷300g,加热至回流,搅拌2小时,tlc检测原料完全消失后,反应液降温至室温,然后把反应液倒入冰水2000ml中,加入有机溶剂二氯甲烷700ml,萃取三次,合并有机相,然后浓缩,通过柱层析分离得到纯净的2-氯-4-(吡啶-3-基)吡啶155g,1hnmr(400mhz,cdcl3):9.11(s,1h),8.85-8.83(m,1h),8.79(d,j=4.0hz,1h),8.55-8.54(m,1h),7.92(d,j=24.0hz,2h),7.43(d,j=8.0hz,1h)。
实施例11
将2-氯-4-(吡啶-3-基)吡啶19g溶于n,n-二甲基甲酰胺200ml中,配置成溶液a;把
实施例12
我们采用计算机药物辅助设计的方法,把所得的化合物分子与肿瘤细胞的靶点蛋白进行分子对接,靶点序号为1m17。我们发现化合物可以很好的进入靶点蛋白口袋里面,化合物分子被靶点蛋白残基氨基酸所包围,与靶点形成非常强的作用效果,其中化合物分子可以分别与天冬氨酸asp831,甲硫氨酸met769形成氢键作用,与苯丙氨酸phe699形成π-π共轭作用。具体见图1,为药物分子和肿瘤靶点对接图。
实施例13
抗肿瘤活性测试
收集生长期肺癌细胞a549,以mts法测定一下化合物的抗癌活性,将细胞以适当浓度(每毫升5×104个细胞)加到96孔细胞培养板中(含10%胎小牛血清得培养液配成单个细胞悬液),培养24小时后,在37℃、体积浓度为5%的co2条件下与不同浓度的化合物作用72小时,然后将mts(最终质量浓度2mg/ml)和dms(最终摩尔浓度30μm)的混合物直接加入含细胞的培养基中,继续置培养箱孵育4h。作用4h后,弃去上清液,每孔加入dmso150μl,振荡,细胞存活率通过其对mts作用的代谢物在酶联免疫监测仪490nm波长下的吸收率测定,药物分子
以上实施例描述了本发明的基本原理、主要特征及优点,本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
- 还没有人留言评论。精彩留言会获得点赞!