一种含三氟甲基3,6’-非对称双吲哚化合物及其合成方法与流程
本发明属于化学合成技术领域,具体涉及一种含三氟甲基3,6’-非对称双吲哚化合物及其合成方法。
背景技术:
吲哚类化合物是一类重要的生物碱,广泛存在于天然产物中。双吲哚类化合物作为吲哚类化合物的一个重要分支,其结构含有两个吲哚并具有吲哚类化合物的一系列特性,在医药以及材料等领域应用广泛。例如,天然产物mucronatinsa和b表现出乙酰胆碱酯酶抑制活性;从新喀里多尼亚海绵中分离出的双吲哚生物碱c和d表现出抗血清素活性,对丝氨酸生长抑制素和神经肽y受体具有很强的活性。目前,对称双吲哚类化合物的合成已被广泛探索,并应用于很多领域。与之相比,非对称双吲哚类化合物其药理特征也引起极大关注,但是关于其合成研究报道较少。
目前,主要通过3-吲哚甲醇和不同吲哚的付-克烷基化反应合成非对称双吲哚类化合物(tetrahedron,2005,61(43):10235-10241;greenchemistry,2016,18(4):1032-1037),但是这类方法只适用于合成3,3’-非对称双吲哚化合物。
含氟基团可替代药物分子中的其他基团,能够显著提高药物的药用价值以及性能。含有三氟甲基(-cf3)的化合物在药物、农药、材料以及染料方面得到了广泛应用。例如,治疗精神抑郁的药物prozac、治疗关节炎的药物celebrex和抗艾滋病药物efavirena等都含有三氟甲基。但是,含氟杂环化合物的制备比较困难,有关其合成的研究报道较少。
非对称双吲哚类化合物以及含氟化合物都在医药领域有显著的功效,将三氟甲基引入到双吲哚类化合物中将具有潜在的药用价值。其次,吲哚c-6位置活性比较低而需要借助合适的导向基团和催化剂的作用,对于吲哚c-6位上的c-h键活化或者官能团化的成功例子极少。
目前,对于非对称双吲哚的研究大多局限于3,3’-非对称双吲哚化合物的合成,而对3,6’-非对称双吲哚化合物的合成研究很少,目前仅有一例文献报道可实现合成3,6’-非对称双吲哚(angew.chem.int.ed.2018,57:11004-11008),该文献使用6-吲哚苄醇与吲哚在磷酸催化下付-克烷基化反应得到3,6’-非对称双吲哚,但该方法的原料不易制备,并且合成的产物仅仅具有双吲哚取代的三级碳中心,不能够得到具有季碳中心的双吲哚化合物。
技术实现要素:
本发明的目的在于解决现有技术中在合成含三氟甲基3,6’-非对称双吲哚化合物方面的不足,提供一种含三氟甲基3,6’-非对称双吲哚化合物及其合成方法。该3,6’-非对称双吲哚化合物具有三氟甲基取代的季碳中心,并且制备方法具有底物适用范围广、简便高效等优点。
为实现上述目的,本发明提供了如下技术方案:
一种含三氟甲基3,6’-非对称双吲哚化合物,其结构式如式(i)所示:
其中,r1、r2、r3为各自独立的基团,r1选自卤素原子、甲氧基、甲基、酯基、羟基或烷基中的一种;r2选自苯基、卤素取代苯基、烷基取代苯基或萘基中的一种;r3选自苯基、环烷基或烷基的一种。
上述含三氟甲基3,6’-非对称双吲哚化合物的合成方法:以三氟甲基吲哚甲醇类化合物和2-取代吲哚为原料,在催化剂作用下,在溶剂中进行反应,将反应产物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,即得。
反应式如下:
进一步,所述三氟甲基吲哚甲醇类化合物的结构式如式(ii)所示:
式(ii)中r1、r2同式(i)中r1、r2对应一致。
进一步,所述2-取代吲哚的结构式如式(iii)所示:
式(iii)中r3同式(i)中r3对应一致。
进一步,所述催化剂选自三氯化铁[fecl3]、高氯酸镍[ni(clo4)2]、三氟甲磺酸镓[ga(otf)3]、三氟甲磺酸钇[y(otf)3]、三氟甲磺酸铟[in(otf)3]或三氟甲磺酸钪[sc(otf)3]中的任意一种,优选为三氟甲磺酸镓,催化效果最好。
进一步,所述溶剂为乙腈、二氯乙烷、甲苯、四氢呋喃、甲基叔丁基醚、硝基甲烷或乙二醇二甲醚中的任意一种,优选为乙腈,产率最佳。
进一步,所述三氟甲基吲哚甲醇类化合物与2-取代吲哚的摩尔比是1:1.5。
进一步,所述催化剂与三氟甲基吲哚甲醇类化合物的摩尔比为(0.01-0.1):1。
进一步,所述反应温度为80℃,反应时间为24-48小时。
作为本发明所述的含三氟甲基3,6’-非对称双吲哚化合物的合成方法的一种优选方案,所述三氟甲基吲哚甲醇类化合物在乙腈中的浓度是0.1mol/l。
本发明的有益效果:
(1)本发明提供了一种含三氟甲基3,6’-非对称双吲哚化合物,并成功实现了它的合成。
(2)本发明以吲哚类化合物为原料,适用底物范围较广,各种官能团耐受性相对较好。
(3)本发明的合成方法操作简便,产率较高。
(4)本发明的含三氟甲基3,6’-非对称双吲哚化合物具有反应活性,具有潜在的生物活性和药用价值。
附图说明
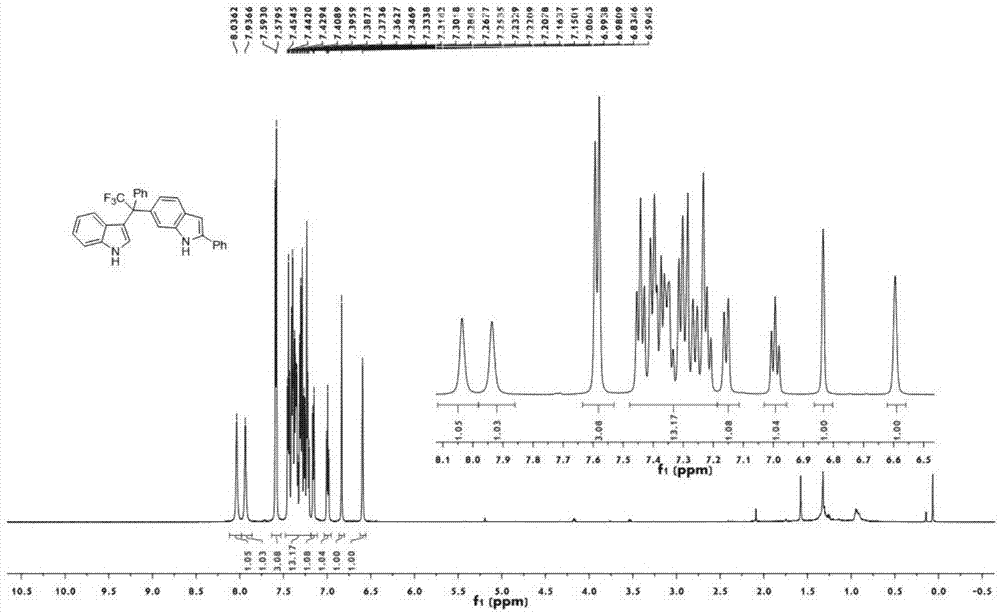
图1为实施例1制得的含三氟甲基3,6’-非对称双吲哚化合物1aa的核磁氢谱图;
图2为实施例1制得的含三氟甲基3,6’-非对称双吲哚化合物1aa的核磁碳谱图;
图3为实施例1制得的含三氟甲基3,6’-非对称双吲哚化合物1aa的核磁氟谱图;
图4为实施例1制得的含三氟甲基3,6’-非对称双吲哚化合物1aa的单晶衍射图。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例对本发明的具体实施方式做详细的说明。
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
实施例1:
反应式为:
含三氟甲基3,6’-非对称双吲哚化合物的合成方法:取25ml圆底烧瓶,依次加入三氟甲基吲哚苄醇2a(0.3mmol)、2-苯基吲哚3a(0.45mmol)、三氟甲磺酸镓(0.03mmol)和乙腈(3毫升),在80℃下搅拌48小时,然后对反应混合物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,所用洗脱剂为乙酸乙酯:石油醚=6:100,分离得到目标产物1aa(120.4mg,白色固体,产率86%)。
图1为本发明实施例1化合物1aa的核磁氢谱图;图2为本发明实施例1化合物1aa的核磁碳谱图;图3为本发明实施例1化合物1aa的核磁氟谱图;图4为本发明实施例1化合物1aa的单晶衍射图。
1hnmr(600mhz,cdcl3):δ8.03(s,1h),7.93(s,1h),7.58(d,j=8.1hz,3h),7.50-7.18(m,13h),7.15(d,j=8.2hz,1h),6.99(t,j=7.6hz,1h),6.83(s,1h),6.59(s,1h).
13cnmr(150mhz,cdcl3):δ140.5,138.9,136.7,136.5,133.9,132.2,129.8,129.0,128.3,128.0,127.9,127.5,127.1,126.5,125.2,122.7,122.2,120.0,199.9;116.6,112.9,111.25,99.5,60.8(q,j=25.2hz).
实施例2:
反应式为:
含三氟甲基3,6’-非对称双吲哚化合物的合成方法:取25ml圆底烧瓶,依次加入三氟甲基吲哚苄醇2b(0.5mmol)、2-苯基吲哚3a(0.75mmol)、三氟甲磺酸镓(0.05mmol)和乙腈(3毫升),80℃下搅拌48小时。然后对反应混合物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,所用洗脱剂为乙酸乙酯:石油醚=6:100,分离得到目标产物1ba(234.1mg,白色固体,产率81%)。
1hnmr(600mhz,dmso-d6):δ11.58(s,1h),11.53(s,1h),7.84(d,2h,j=7.6hz),7.56(d,1h,j=8.5hz),7.50(d,1h,j=8.6hz),7.46-7.41(m,5h),7.32-7.28(m,3h),7.20(s,1h),7.11(d,1h,j=8.3hz),7.02(d,1h,j=8.3hz),6.94(s,1h),6.87(s,1h),6.73(s,1h).
13cnmr(150mhz,dmso-d6):δ140.2,139.3,137.2,135.9,132.5,132.4,129.6,129.5,129.4,128.7,128.3,128.3,128.0,127.4,125.4,124.1,121.8,121.1,120.1,114.4,114.1,113.0,98.8,60.5(q,j=24.0hz).
实施例3:
反应式为:
含三氟甲基3,6’-非对称双吲哚化合物的合成方法:取25ml圆底烧瓶,依次加入三氟甲基吲哚苄醇2c(0.2mmol)、2-苯基吲哚3a(0.3mmol)、三氟甲磺酸镓(0.02mmol)和乙腈(3毫升),80℃下搅拌24小时。然后对反应混合物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,所用洗脱剂为乙酸乙酯:石油醚=20:100,分离得到目标产物1ca(117.3mg,白色固体,产率90%)。
1hnmr(600mhz,cdcl3)δ=8.70(s,1h),8.39(s,1h),7.64(d,2h,j=3.0hz),7.59(d,1h,j=8.5hz),7.52(s,1h),7.46-7.43(m,3h),7.40-7.32(m,6h),7.28(s,1h),7.25(m,6h),7.17(d,1h,j=8.5hz),6.85(d,1h,j=1.4hz),6.82(d,1h,j=2.6hz).
13cnmr(150mhz,cdcl3)δ139.7,139.3,138.6,136.6,133.1,132.1,129.6,129.4,129.1,128.6,128.3,128.1,128.0(d,j=6.7hz),126.17,125.27,125.04,121.90,120.80,120.2,117.6,112.51,112.5,102.9,99.5,60.5(q,j=24.2hz).
实施例4:
反应式为:
含三氟甲基3,6’-非对称双吲哚化合物的合成方法:取25ml圆底烧瓶,依次加入三氟甲基吲哚苄醇2d(0.5mmol)、2-苯基吲哚3a(0.75mmol)、三氟甲磺酸镓(0.05mmol)和乙腈(3毫升),80℃下搅拌48小时。然后对反应混合物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,所用洗脱剂为乙酸乙酯:石油醚=6:100,分离得到目标产物1da(139.4mg,白色固体,产率81%)。
1hnmr(600mhz,cdcl3):δ8,25(s,1h),8.06(s,1h),7.63(d,2h,j=7.8hz),7.59(d,1h,j=8.5hz),7.46-7.41(m,4h),7.35-7.33(m,4h),7.29(d,2h,j=8.8hz),7.25(d,1h,j=8.6hz),6.86-6.83(m,2h),6.76(s,1h),6.38(s,1h),3.50(s,3h).
13cnmr(150mhz,cdcl3):δ153.8,140.4,138.9,136.6,133.8,132.2,131.7,129.9,129.0,128.3,128.0,127.8,127.5,127.2,127.1,125.1,122.3,119.9,116.1,112.8,112.5,111.7,104.1,99.5,61.4(q,j=24.8hz),55.51.
实施例5:
反应式为:
含三氟甲基3,6’-非对称双吲哚化合物的合成方法:取25ml圆底烧瓶,依次加入三氟甲基吲哚苄醇2e(0.3mmol)、2-苯基吲哚3a(0.45mmol)、三氟甲磺酸镓(0.03mmol)和乙腈(3毫升),80℃下搅拌48小时。然后对反应混合物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,所用洗脱剂为乙酸乙酯:石油醚=6:100,分离得到目标产物1ea(127.5mg,白色固体,产率80%)。
1hnmr(400mhz,dmso-d6):δ16.32(s,1h),16.10(s,1h),12.60-12.54(m,3h),12.44-12.31(m,4h),12.24-12.17(m,3h),12.01(t,1h,j=7.4hz),11.95(s,1h),11.86(t,1h,j=18.6hz),11.77(d,1h,j=8.7hz),11.69(d,1h,j=1.4hz),11.63-11.53(m,3h).
13cnmr(100mhz,dmso-d6):δ146.6,144.2,142.2,141.9,138.7,137.2,136.8,134.7,134.3,134.1,133.9,133.2,132.8,132.6,130.8,130.6,130.5,130.2,129.9,127.9,126.6,126.0,125.8,125.0,124.4,118.5,117.8,117.3,103.6,65.5(q,j=24.4hz).
实施例6:
含三氟甲基3,6’-非对称双吲哚化合物的合成方法:取25ml圆底烧瓶,依次加入三氟甲基吲哚苄醇2f(0.5mmol)、2-苯基吲哚3a(0.75mmol)、三氟甲磺酸镓(0.05mmol)和乙腈(3毫升),80℃下搅拌48小时。然后对反应混合物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,所用洗脱剂为乙酸乙酯:石油醚=8:100,分离得到目标产物1fa(176.1mg,白色固体,产率73%)。
1hnmr(600mhz,cdcl3):δ7.90(1s,1h),7.80(1s,1h),7.61(d,1h,j=8.5hz),7.55(d,2h,j=7.3hz),7.45(t,2h,j=7.6hz),7.38-7.24(m,8h),7.13(t,2h,j=8.2hz),7.04(t,1h,j=8.1hz),6.86(s,1h),6.54(s,1h),2.42(s,3h).
13cnmr(150mhz,cdcl3)δ138.9,137.5,137.3,136.8,136.5,134.1,132.1,129.7,129.4,129.1,128.9,128.2,127.9,127.5,127.2,126.6,125.3,122.7,122.2,120.0,119.9,116.7,113.0,111.4,99.5,60.5(q,j=25.0hz),21.1.
实施例7:
含三氟甲基3,6’-非对称双吲哚化合物的合成方法:取25ml圆底烧瓶,依次加入三氟甲基吲哚苄醇2g(0.5mmol)、2-苯基吲哚3a(0.75mmol)、三氟甲磺酸镓(0.05mmol)和乙腈(3毫升),80℃下搅拌48小时。然后对反应混合物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,所用洗脱剂为乙酸乙酯:石油醚=6:100,分离得到目标产物1ga(129.7mg,白色固体,产率50%)。
1hnmr(400mhz,cdcl3):δ7.96(s,1h),7.89(s,1h),7.79(t,2h,j=8.0hz),7.69(d,1h,j=8.8hz),7.57-7.45(m,6h),7.41-7.17(m,9h),6.93(t,1h,j=8.0hz),6.79(d,1h,j=1.32hz),6.48(d,1h,j=2.5hz).
13cnmr(100mhz,cdcl3)δ139.0,138.0,136.78,136.5,133.9,132.8,132.5,132.1,129.0,128.7,128.4,127.9,127.7,127.4,127.3,126.5,126.5,126.1,125.2,122.7,122.2,120.0,119.9,116.5,112.9,111.3,99.5,61.0(q,j=24.9hz).
实施例8:
含三氟甲基3,6’-非对称双吲哚化合物的合成方法:取25ml圆底烧瓶,依次加入三氟甲基吲哚苄醇2a(0.3mmol)、2-苯基吲哚3b(0.45mmol)、三氟甲磺酸镓(0.03mmol)和乙腈(3毫升),80℃下搅拌36小时。然后对反应混合物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,所用洗脱剂为乙酸乙酯:石油醚=6:100,分离得到目标产物1ab(130.9mg,白色固体,产率82%)。
1hnmr(400mhz,cdcl3):δ7.53(s,1h),7.43(d,2h,j=8.4hz),7.26-7.08(m,9h),6.96-6.91(m,2h),6.25(s,1h),6.21(s,1h),1.27(s,9h).
13cnmr(100mhz,cdcl3)δ150.1,140.8,136.8,135.5,132.9,129.9,129.8,128.0,127.5,127.4,127.0,126.6,122.8,122.8,122.1,121.5,119.8,119.3,116.8,112.5,111.3,96.7,60.8(q,j=24.9hz),31.9,30.3
实施例9:
含三氟甲基3,6’-非对称双吲哚化合物的合成方法:取25ml圆底烧瓶,依次加入三氟甲基吲哚苄醇2a(0.3mmol)、2-苯基吲哚3c(0.45mmol)、三氟甲磺酸镓(0.03mmol)和乙腈(3毫升),80℃下搅拌24小时。然后对反应混合物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,所用洗脱剂为乙酸乙酯:石油醚=8:100,分离得到目标产物1ac(144.8mg,白色固体,产率80%)。
1hnmr(400mhz,cdcl3):δ7.63(s,1h),7.33(d,1h,j=8.5hz),7.22-7.20(m,10h),7.14-7.03(m,10h),6.88(t,1h,j=6.9hz),6.84(s,1h),6.32(s,1h),5.95(s,1h),5.37(s,1h).
13cnmr(100mhz,cdcl3)δ141.9,140.6,136.7,135.8,133.2,129.9,129.0,129.0,128.6,127.9,127.3,127.3,127.2,126.9,126.5,122.7,122.1,121.6,119.6(d,j=33.5hz),116.7,112.7,111.2,102.4,60.7(q,j=25.0hz),51.1.
实施例10:
含三氟甲基3,6’-非对称双吲哚化合物的合成方法:取25ml圆底烧瓶,依次加入三氟甲基吲哚苄醇2a(0.2mmol)、2-苯基吲哚3c(0.3mmol)、三氟甲磺酸镓(0.02mmol)和乙腈(3毫升),80℃下搅拌36小时。然后对反应混合物进行旋蒸浓缩,再通过200~300目硅胶柱层析分离,所用洗脱剂为乙酸乙酯:石油醚=6:100,分离得到目标产物1ad(59.4mg,白色固体,产率63%)。
1hnmr(600mhz,cdcl3)δ7.86(s,1h),7.50(d,1h,j=8.5hz),7.34-7.17(m,9h),7.01(t,1h,j=9.0hz),6.96(s,1h),6.33(s,1h),6.24(s,1h),2.58(s,1h),2.00(s,2h),1.87(s,2h),1.78(d,1h,j=12.8hz),1.44-1.40(m,4h),1.35-1.32(m,3h).
13cnmr(150mhz,cdcl3)δ146.3,140.8,136.6,135.0,132.4,129.8,129.3,127.9,127.5,127.4,127.3,126.5,122.8,122.1,121.3,119.8,119.2,116.8,112.5,111.2,96.9,60.7(q,j=25.5hz),37.3,32.9(d,j=7.5hz),26.2(d,j=25.5hz).
由此可见,本发明所提供的一种含三氟甲基3,6’-非对称双吲哚化合物的合成方法可以实现吲哚c-6位置的官能团化,整个反应利用一锅法方式进行,操作简便,产率高并且反应底物范围广,研发制得的含三氟甲基3,6’-非对称双吲哚化合物具有潜在生物活性,可经过后续测试或者改性成为药物。
应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!
- 含离子液体共溶剂介质中制备(R)‑3,5‑双三氟甲基苯乙醇的方法与流程
- 一种邻二氟苯的合成方法与制造工艺
- 2,2,2‑三氟乙硫醇的合成的制造方法与工艺
- 2-氯-3-(甲基硫烷基)-N-(1-甲基-1H-四唑-5-基)-4-(三氟甲基)苯甲酰胺或其盐在耐受HPPD抑制剂除草剂的转基因作物植物的区域中防治不想要的植物的用途的制造方法与工艺
- (R)‑3,5‑双(三氟甲基)苯乙醇的生物制备方法与制造工艺
- 2-(2-吡啶)-6-(2-氯-3-三氟甲基苯甲酰基)-5,7,8-三氢吡啶并[4,3-d]嘧啶及其制备方法与制造工艺
- 一种1,3,4-噻二唑类化合物及其制备方法与制造工艺
- 一种1,3,4-噻二唑类化合物及其制备方法与制造工艺
- 一种阿瑞吡坦中间体及其制备方法与制造工艺
- 一类含4,4’‑二氟二苯甲基的不对称α‑二亚胺镍配合物、其中间体、制备方法及其应用与制造工艺