一种吸水性树脂的反相悬浮聚合制备工艺的制作方法
本发明属于高分子材料领域,涉及一种吸水性树脂的反相悬浮聚合制备工艺,具体涉及一种采用先聚合、后中和的反相悬浮聚合工艺制备高吸水性树脂(sap,superabsorbentpolymer)的方法。
背景技术:
高吸水性树脂是一种轻度交联的高分子聚合物,由于其强大的吸水与保水能力,被广泛用于纸尿裤、卫生巾等的卫生材料领域、电缆阻水材料以及农林、园艺保水剂等。
作为卫生材料中使用的吸水性树脂,已知有聚丙烯酸部分中和物、淀粉-丙烯酸接枝聚合物的中和物、淀粉-丙烯腈接枝聚合物的水解物、乙酸乙烯酯-丙烯酸酯共聚物的皂化物等。
目前吸水性树脂主要采用水溶液聚合法与反相悬浮(乳液)聚合法制备,约有95%的高吸水性树脂采用水溶液聚合法制备,代表厂家有日本触媒、basf、赢创德固赛、三大雅、lg等;约有5%的采用反相悬浮聚合法制备,代表厂家是日本住友精化与三菱化学。
作为吸水性树脂的制备工艺,绝大部分是采用先中和后聚合的制备工艺:即直接将丙烯酸类单体与氢氧化钠等碱液经过酸碱中和反应,得到所谓的丙烯酸中和液,再加入交联剂和引发剂,然后进行高温下的自由基聚合得到吸水性树脂,比较典型的有日本触媒(例如,cn102549028a、cn102482433a、cn101177462a、cn1784430a),住友精化(例如,cn1337977a、cn1053796a、cn1175962a),三菱化学(例如,cn1146997a、cn1197804a)等;而也有一小部分是采用先聚合后中和的制备工艺:即在低温下将包含引发剂与交联剂的丙烯酸溶液直接进行自由基聚合,聚合完成之后,再加入氢氧化钠等碱液中和掉聚丙烯酸中的部分羧基,也可以得到吸水性树脂,比较典型的有三大雅(例如,cn1589304a、cn106459598a、cn107428948a、cn107406595a),诺尔(例如,cn106987075a)等。
先中和后聚合的制备工艺和先聚合后中和的制备工艺各有各的优势,由于丙烯酸单体的聚合热较高,直接聚合对反应条件要求比较苛刻,因此需要在低温下或者散热状态良好的条件下进行聚合,而配置成丙烯酸中和液的话,其反应速率常数下降,使得聚合条件变得温和、可控。
水溶液聚合法中采用先聚合、后中和的生产工艺有一定的缺陷,比如酸碱中和过程中大量的中和热需要移去,造成能源的浪费;并且酸碱中和过程中由于温升明显,造成得到的丙烯酸中和液中丙烯酸二聚体的含量急剧增加,这使得中和液的存储时间受到限制,并且还会进一步影响吸水性树脂的最终的产品性能。
反相悬浮聚合作为一种独特的吸水性树脂的制备工艺,可以制备高吸水速率的吸水性树脂,极大的满足了复合型芯体市场对超薄性的追求。目前,只有住友精化具备大规模工业化制造的能力,但是其制备方式也是采用先中和、后聚合的工艺。本领域还不存在将先聚合、后中和的工艺运用到反相悬浮聚合制备方式中。
技术实现要素:
本发明的目的是改进现有技术中生产吸水性树脂的各类工艺中存在的缺陷,提供一种新的制备差异化吸水性树脂的方法。
为了解决以上技术问题,在本发明的一个方面,提供一种采用先聚合、后中和的反相悬浮聚合工艺制备吸水性树脂的方法,包括以下步骤:
a.一段聚合:向溶有分散剂的石油烃类溶剂中,加入包含一段引发剂、交联剂以及共聚单体的一段(甲基)丙烯酸水溶液,进行一段油包水反相悬浮聚合,得到含有一段聚(甲基)丙烯酸胶粒的悬浮液;
b.二段聚合:向步骤a得到的所述含有一段聚(甲基)丙烯酸胶粒的悬浮液中加入包含表面活性剂、二段引发剂、交联剂以及共聚单体的二段(甲基)丙烯酸水溶液,使所述二段(甲基)丙烯酸水溶液的(甲基)丙烯酸液滴与一段聚(甲基)丙烯酸胶粒彼此吸附并聚集,进行二段油包水反相悬浮聚合,得到含有二段聚(甲基)丙烯酸胶粒的悬浮液;
所述表面活性剂的亲水亲油平衡值hlb>7;
c.酸碱中和:向步骤b得到的所述含有二段聚(甲基)丙烯酸胶粒的悬浮液中加入碱性溶液,使之与二段聚(甲基)丙烯酸胶粒发生酸碱中和反应;
d.共沸脱水与表面交联:将步骤c得到的悬浮液升温至水与石油烃类溶剂的共沸点(共沸点t<100℃),将悬浮液中70-90%的水移除,添加表面交联剂进行表面交联;
e.溶剂脱除与干燥:采用过滤或者蒸馏工艺去除体系中剩余的水和石油烃类溶剂,最后干燥以得到吸水性树脂;
所述(甲基)丙烯酸为丙烯酸或甲基丙烯酸。
在一个优选的实施方案中,上述方法中,步骤a中所述石油烃类溶剂选自环戊烷、环己烷、正庚烷、正己烷和正辛烷中的一种或多种,优选正庚烷和/或环己烷;
步骤a中所述分散剂选自蔗糖脂肪酸酯、失水山梨醇单硬脂酸酯、失水山梨醇单油酸酯、三聚甘油单硬脂酸酯、十八烷基单磷酸酯和马来酸酐改性的乙烯-丙烯共聚物中的一种或多种;
步骤a中所述一段引发剂为热引发剂,选自过硫酸钠、过硫酸钾、过硫酸铵、2,2'-偶氮二异丙基咪唑林盐酸盐(v-50)、2,2'-偶氮(2-甲基-n-(2-羟基乙基)丙酰胺)(va-086)和2,2-氮杂双(2-咪唑林)二盐酸盐(va-044)中的一种或多种;
步骤a中所述交联剂选自乙二醇二缩水甘油醚、n,n'-亚甲基双丙烯酰胺、聚乙二醇双丙烯酸酯和季戊四醇三烯丙基醚中的一种或多种;
步骤a中所述共聚单体选自丙烯酰胺、甲基丙烯酰胺、醋酸乙烯酯、丙烯酸缩水甘油醚、甲基丙烯酸缩水甘油醚、丙烯酸羟乙酯、甲基丙烯酸羟乙酯和醋酸乙烯酯中的一种或多种。
在一个优选的实施方案中,上述任一所述的方法中,步骤a中所述石油烃类溶剂的用量为所述一段(甲基)丙烯酸水溶液质量的100-500%,优选为100-300%;
步骤a中所述分散剂的用量为所述石油烃类溶剂质量的0.1-10%,优选为0.2-1%;
步骤a中所述一段引发剂的用量为一段(甲基)丙烯酸质量的0.005-5%,优选为0.02-2%;
步骤a中所述交联剂的用量为一段(甲基)丙烯酸质量的0.001-1%,优选为0.01-0.5%;
步骤a中所述共聚单体的用量为一段(甲基)丙烯酸质量的0.005-5%,优选为0.02-2%;
步骤a中所述一段(甲基)丙烯酸水溶液中的一段(甲基)丙烯酸的质量浓度为10-60%,优选为20-40%;
所述一段(甲基)丙烯酸为所述一段(甲基)丙烯酸水溶液中的(甲基)丙烯酸;
步骤a中所述一段油包水反相悬浮聚合(高温聚合)为高温热引发,聚合温度为40-80℃,优选为60-80℃,时间为0.5-3h,优选为0.8-2.5h。
在一个优选的实施方案中,上述方法中,步骤b中所述表面活性剂选自烷基酚聚氧乙烯醚(op-10)、失水山梨糖醇月桂酸酯(span-20)、聚氧乙烯脱水山梨糖醇单油酸酯(tween-80)、蔗糖脂肪酸酯(s-970)、聚乙烯醇(pva)和聚氧乙烯十二烷基醚磷酸盐中的一种或多种;
步骤b中所述二段引发剂为氧化还原引发剂,选自过硫酸钠/亚硫酸钠、过硫酸钠/亚硫酸氢钠、叔丁基过氧化氢/硫酸亚铁和过氧化氢/l-抗坏血酸中的一种或多种;
步骤b中所述交联剂选自乙二醇二缩水甘油醚、n,n'-亚甲基双丙烯酰胺、聚乙二醇双丙烯酸酯和季戊四醇三烯丙基醚中的一种或多种;
步骤b中所述共聚单体选自丙烯酰胺、甲基丙烯酰胺、醋酸乙烯酯、丙烯酸缩水甘油醚、甲基丙烯酸缩水甘油醚、丙烯酸羟乙酯、甲基丙烯酸羟乙酯和醋酸乙烯酯中的一种或多种。
在一个优选的实施方案中,上述任一所述的方法中,步骤b中所述表面活性剂的用量为一段(甲基)丙烯酸和二段(甲基)丙烯酸中(甲基)丙烯酸总质量的0.01-10%,优选为0.1-1.5%;
步骤b中所述二段引发剂的用量为二段(甲基)丙烯酸质量的0.005-5%,更优选为0.02-2%;
步骤b中所述交联剂的用量为二段(甲基)丙烯酸质量的0.001-1%,优选为0.01-0.5%;
步骤b中所述共聚单体的用量为二段(甲基)丙烯酸质量的0.005-5%,优选为0.02-2%;
步骤b中所述二段(甲基)丙烯酸水溶液中二段(甲基)丙烯酸的质量浓度为10-60%,优选为20-45%;
所述一段(甲基)丙烯酸为所述一段(甲基)丙烯酸水溶液中的(甲基)丙烯酸;所述二段(甲基)丙烯酸为所述二段(甲基)丙烯酸水溶液中的(甲基)丙烯酸;
步骤b中所述二段油包水反相悬浮聚合(低温聚合)为氧化还原引发,聚合温度为20-60℃,优选为30-50℃,时间为0.5-3h,优选为0.5-2h;
本发明中从分散剂开始加入到聚合过程结束均在搅拌下进行,所用搅拌转速为200-600rpm/min。
在一个优选的实施方案中,上述方法中,步骤c中所述碱性溶液选自氢氧化钠、氢氧化钾、碳酸氢钠、碳酸钠、碳酸钾和碳酸铵水溶液中的一种或多种,优选氢氧化钠、氢氧化钾和碳酸氢钠水溶液中的一种或多种。
在一个优选的实施方案中,上述任一所述的方法中,步骤c中所述碱性溶液的质量浓度为20-55%;
步骤c中所述酸碱中和反应的中和度为60-90%,优选为70-80%;
步骤c中发生所述酸碱中和反应的温度为30-60℃,优选为40-50℃,时间为10-90min,优选为10-30min。
在一个优选的实施方案中,上述方法中,步骤d中所述表面交联剂为含有至少两个能与羧基反应的官能团的化合物,优选多缩水甘油基化合物、多元醇和碳酸亚烷基酯中的一种或多种,更优选乙二醇二缩水甘油醚、甘油二缩水甘油醚、聚乙二醇二缩水甘油醚、甘油、丙二醇、乙二醇、1,4-丁二醇、碳酸亚乙酯和碳酸亚丙酯中的一种或多种。
在一个优选的实施方案中,上述任一所述的方法中,步骤d中所述表面交联剂的用量为一段(甲基)丙烯酸和二段(甲基)丙烯酸总质量的0.001-15%,优选0.01-10%;
所述一段(甲基)丙烯酸为所述一段(甲基)丙烯酸水溶液中的(甲基)丙烯酸;所述二段(甲基)丙烯酸为所述二段(甲基)丙烯酸水溶液中的(甲基)丙烯酸;
步骤d中所述表面交联的温度为60-100℃,优选为70-90℃,时间为0.5-3h,优选为1-2h;
步骤e中所述蒸馏的温度为100-150℃,时间为0.2-1h;
步骤e中所述干燥的温度为100-180℃,时间为0.5-2h;
所述吸水性树脂的最终含水量为1-10%,优选为2-5%。
在本发明的另一方面,还提供由上述任一项所述的方法制备得到的吸水性树脂。
与传统的先中和、后聚合的吸水性树脂的制备工艺不同,本发明采用的是先聚合、后中和的高吸水性树脂制备工艺,其直接将丙烯酸水溶液进行一段反相悬浮聚合(高温热引发),聚合结束后,将温度降至水溶性引发剂分解温度以下,再加入包含表面活性剂的二段丙烯酸水溶液进行二段反相悬浮聚合(低温氧化还原引发),边进行颗粒团聚边进行二段聚合;聚合结束后加入适量的碱性溶液与得到的聚丙烯酸或聚甲基丙烯酸发生酸碱中和反应,有效的利用了聚合过程的温度和热量,得到吸水性树脂水凝胶颗粒。最后,对得到的含水凝胶粒子共沸脱水、表面交联、蒸馏、干燥和筛分即可得到目标高吸水性树脂颗粒。最终sap产品的性能可通过后期加入碱的用量来调整。
本发明制备的sap兼有较好的通液速率和1min纯水吸水量,推测可能是高低温引发搭配,聚合过程比较平稳,并且交联网络结构比较均匀,使得到的sap颗粒的吸水后凝胶强度增加而导致的。
本发明丰富了吸水性树脂的制备工艺,并为具有差异化性能的吸水性树脂的开发提供了一种思路。
附图说明
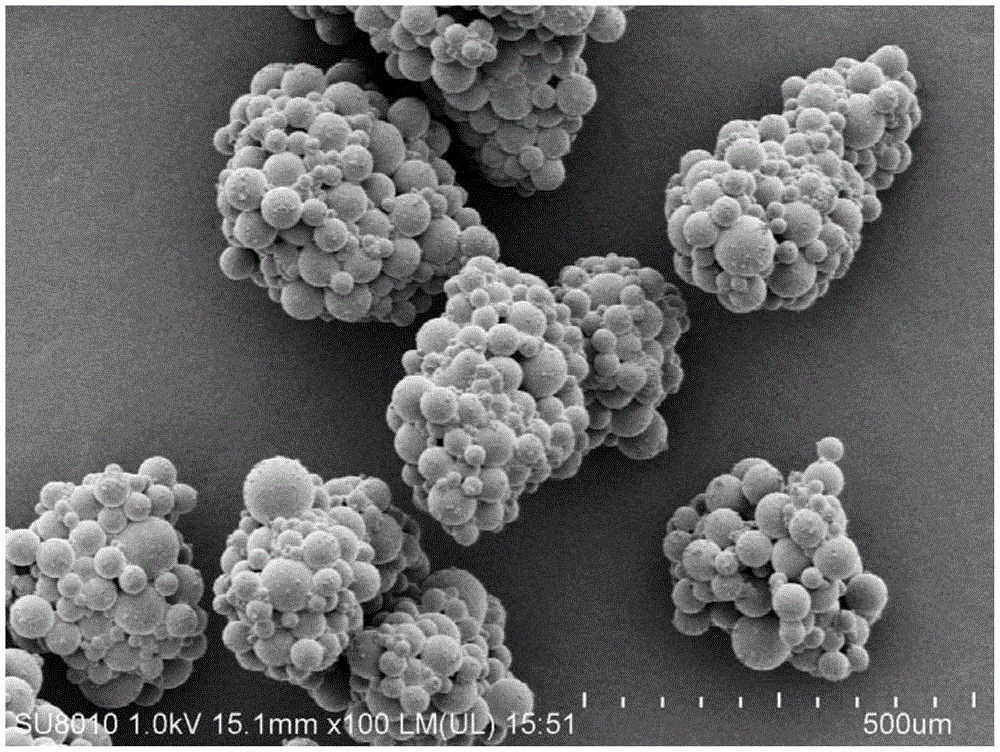
图1为本发明实施例1中所得到的吸水性树脂的sem(扫描电子显微镜)图像。
具体实施方式
下述实施例中所使用的实验方法中所涉及的某些具体步骤,如无特殊说明,均为常规操作。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
以下结合具体实施例,对本发明作进一步说明。应理解,以下实施例仅用于说明本发明而非用于限定本发明的范围。
本发明的高吸水性树脂的基本性能指标测试方法如下:
吸液倍率、离心保水与加压吸液倍率测试方法执行gb/t22875-2018和iso17190-5-2001标准,其余物理性质按照如下方法进行测试:
通液速率(g/min)
自制通液性测量装置(采用内径25mm、外径31mm、高35cm的塑料圆筒,底面贴有63μm尼龙网)。取0.1g高吸水性树脂,倒入100ml烧杯中,加入40ml生理盐水使其膨润。30min后,将膨润的凝胶全部倒入测量装置内(烧杯中如有残留,用生理盐水冲洗至全部倒入)。将100g砝码缓慢压在前述测量装置内的凝胶上,之后加入适量生理盐水至测试刻度线(约28cm处),将测量装置垂直悬空,此时会有液滴缓慢流下,测量1min内通过凝胶的生理盐水量g/min,即为通液速率,此数值越高,说明sap吸液后的扩散传导性越好。
可萃取物含量(%)
用分析天平(精确度:0.0001g)准确称取1.000±0.005gsap,放入250ml烧杯中,加入200ml生理盐水(质量浓度0.9%),放入磁力转子(长度约3cm)后,用封口膜密封,置于磁力搅拌器上进行搅拌(转速约为500rpm),16小时后,停止搅拌,静置10分钟,将上层清液用滤纸过滤出50ml滤液。
用0.1mol/lnaoh溶液将滤液滴定至ph=10,消耗溶液体积记为[naoh],然后使用0.1mol/l盐酸溶液滴定至ph=2.7,消耗溶液体积记为[hcl]。同时取蒸馏水进行空白实验,消耗两种溶液的体积记为[bnaoh],[bhcl]。
可萃取物含量越低,说明吸水性树脂低分子量部分越少。
1min纯水吸收量(g/g)
向盛有250ml纯水的烧杯中,加入0.5g(精确至0.01g)烘干的高吸水性树脂样品,并开始用秒表计时,1min时,将该烧杯内的高吸水性树脂样品和水搅拌均匀并立即导入滤袋中,稍微将自由水滴净后,称量滤袋的重量m2,将高吸水性树脂样品从滤袋中取出称量滤袋的重量m1,则:高吸水性树脂样品的1min纯水吸收量(g/g)=(m2-m1)/0.5,此数值反映了sap在单位时间内的纯水吸收量,数值越高,说明吸水速率越快。
蔗糖脂肪酸酯(s-370)为三菱化学(上海)有限公司产品,cas号:37318-31-3。
马来酸酐改性的乙烯-丙烯共聚物(1105a)为三井化学株式会社产品,cas号:31069-12-2。
2,2'-偶氮二异丙基咪唑林盐酸盐(v-50)为wako(日本和光纯药株式会社)公司产品,cas号:2997-92-4。
乙二醇二缩水甘油醚为上海长濑贸易有限公司产品,cas号:2224-15-9。
烷基酚聚氧乙烯醚(op-10)为南京古田化工有限公司产品,cas号:9036-19-5。
失水山梨糖醇月桂酸酯(span20)为北京百灵威科技有限公司产品,cas号:1338-39-2。
失水山梨醇单硬脂酸酯(span60)为国药集团化学试剂有限公司产品,cas号:1338-41-6。
竞品a为日本三大雅高分子株式会社产品,水溶液聚合工艺制备:先聚合、后中和;竞品b为株式会社日本触媒产品,水溶液聚合工艺制备:先中和、后聚合;竞品c为住友化学株式会社产品,反相悬浮聚合工艺制备:先中和、后聚合;该三种竞品的所用基本原料如表1所示。
表1三种竞品所用基本原料
实施例1:
1、一段聚合
将240g正庚烷(155.0wt%,基于一段丙烯酸水溶液的质量)加入到装有搅拌器、回流冷凝器、温度计和氮气入口管的1l四口圆底烧瓶中。向其中加入0.92g蔗糖脂肪酸酯(s-370)(0.38wt%,基于正庚烷的质量),升温至60℃并以250rpm的搅拌速度使其均匀溶解分散,然后降温至40℃。将溶有0.055g过硫酸钠(0.10wt%,基于一段丙烯酸的质量)、0.028g2,2'-偶氮二异丙基咪唑林盐酸盐(v-50)(0.05wt%,基于一段丙烯酸的质量)、0.278g甲基丙烯酸羟乙酯(0.5wt%,基于一段丙烯酸的质量)、22.1mg乙二醇二缩水甘油醚(0.04wt%,基于一段丙烯酸的质量)的水溶液99.7g加入到55.2g丙烯酸(即,一段丙烯酸)中,得到一段丙烯酸水溶液(一段丙烯酸的质量浓度为35.6%)。将该一段丙烯酸水溶液加入到上述四口圆底烧瓶中,边搅拌边通入氮气以充分除氧。然后升温至60℃反应2h进行一段油包水(w/o)反相悬浮聚合,得到含有一段聚丙烯酸胶粒的悬浮液。
2、二段聚合
将溶有0.055g过硫酸钠(0.1wt%,基于二段丙烯酸的质量)、0.278g甲基丙烯酸羟乙酯(0.5wt%,基于二段丙烯酸的质量)、22.1mg乙二醇二缩水甘油醚(0.04wt%,基于二段丙烯酸的质量)和0.55g烷基酚聚氧乙烯醚(op-10)(hlb为13.5)(0.5wt%,基于一段和二段丙烯酸的总质量)的水溶液68.8g加入到55.2g丙烯酸(即,二段丙烯酸)中,然后加入0.022g亚硫酸钠(0.04wt%,基于二段丙烯酸的质量)以得到二段丙烯酸水溶液(二段丙烯酸的质量浓度为44.4%)。此后,将步骤1得到的含有一段聚丙烯酸胶粒的悬浮液冷却至40℃,加入二段丙烯酸水溶液,氮气充分置换后,在350rpm的转速下搅拌30min,使得二段丙烯酸水溶液的丙烯酸液滴与一段聚丙烯酸胶粒彼此吸附并聚集。然后,边发生颗粒团聚边进行二段油包水反相悬浮聚合,得到含有二段聚丙烯酸胶粒的悬浮液。
3、酸碱中和
二段聚合结束后,将143.8g质量浓度为32%的氢氧化钠水溶液以2-3滴/秒的滴加速度滴入步骤2得到的含有二段聚丙烯酸胶粒的悬浮液。此时,加入的碱液与得到的二段聚丙烯酸胶粒发生酸碱中和反应,酸碱中和反应的温度为40℃,时间为15min,中和度75%,待体系温度恒定后,继续搅拌15min。
4、共沸脱水与表面交联
将步骤3得到的悬浮液升温至100℃进行共沸脱水并使得正庚烷回流,共脱除200g水(75%的水,基于四口圆底烧瓶中总共266g的水)。将体系温度调整为80℃,向悬浮液中加入质量浓度为1%的乙二醇二缩水甘油醚水溶液9.2g(乙二醇二缩水甘油醚用量为一段和二段丙烯酸总质量的0.08%),在此温度下反应1h进行表面交联。
5、溶剂脱除与干燥
将体系升温至120℃,蒸馏0.5h去除体系中剩余的水与正庚烷,此时可以观察到葡萄串状的高吸水性树脂(sap)粒子沉在烧瓶底部,如图1所示。最后,将其在130℃下干燥1h并筛分可得到所需粒径(150-710微米,占85%)的吸水性树脂产品,最终含水量为3%。
实施例2:
除了将二段聚合中使用的0.55g烷基酚聚氧乙烯醚(op-10,hlb=13.5)替换为0.45g失水山梨糖醇月桂酸酯(span20,hlb=8.6)(0.4wt%,基于一段和二段丙烯酸的总质量)外,重复实施例1的操作。最终得到的吸水性树脂的形态与实施例1类似,并且最终含水量为3%。
实施例3:
除了将聚合前的分散剂0.92g蔗糖脂肪酸酯(s-370)(0.38wt%,基于正庚烷的质量)替换为0.46g失水山梨醇单硬脂酸酯(span60,hlb=4.7)(0.19wt%,基于正庚烷的质量)和0.46g马来酸酐改性的乙烯-丙烯共聚物(1105a)(0.19wt%,基于正庚烷的质量),并升温至80℃并以300rpm的搅拌速度使其均匀溶解分散,然后降温至40℃外,重复实施例1的操作。最终得到的吸水性树脂的形态与实施例1类似,并且最终含水量为4%。
实施例4:
除了将一段聚合和二段聚合的共聚单体甲基丙烯酸羟乙酯替换为醋酸乙烯酯以外,并将每段的添加量改为0.552g(1.0wt%,基于每段丙烯酸的质量)外,重复实施例1的操作。最终得到的吸水性树脂的形态与实施例1类似,并且最终含水量为2%。
实施例5:
除了将酸碱中和步骤中143.8g质量浓度为32%的氢氧化钠水溶液替换为197.8g质量浓度为50.5%的碳酸钠水溶液外,重复实施例1的操作。最终得到的吸水性树脂的形态与实施例1类似,并且最终含水量为5%。
实施例6:
1、一段聚合
将240g正庚烷(155.0wt%,基于一段丙烯酸水溶液的质量)加入到装有搅拌器、回流冷凝器、温度计和氮气入口管的1l四口圆底烧瓶中。向其中加入0.92g蔗糖脂肪酸酯(s-370)(0.38wt%,基于正庚烷的质量),升温至60℃并以250rpm的搅拌速度使其均匀溶解分散,然后降温至40℃。将溶有0.055g过硫酸钠(0.10wt%,基于一段丙烯酸的质量)、0.028g2,2'-偶氮二异丙基咪唑林盐酸盐(v-50)(0.05wt%,基于一段丙烯酸的质量)、0.278g甲基丙烯酸羟乙酯(0.5wt%,基于一段丙烯酸的质量)、22.1mg乙二醇二缩水甘油醚(0.04wt%,基于一段丙烯酸的质量)的水溶液99.7g加入到55.2g丙烯酸(即,一段丙烯酸)中,得到一段丙烯酸水溶液(一段丙烯酸的质量浓度为35.6%)。将该一段丙烯酸水溶液加入到上述四口圆底烧瓶中,边搅拌边通入氮气以充分除氧。然后升温至80℃反应0.5h进行一段油包水(w/o)反相悬浮聚合,得到含有一段聚丙烯酸胶粒的悬浮液。
2、二段聚合
将溶有0.092g质量浓度为30%的h2o2(h2o2为0.05wt%,基于二段丙烯酸的质量)、0.278g甲基丙烯酸羟乙酯(0.5wt%,基于二段丙烯酸的质量)、22.1mg乙二醇二缩水甘油醚(0.04wt%,基于二段丙烯酸的质量)和0.55g烷基酚聚氧乙烯醚(op-10)(hlb为13.5)(0.5wt%,基于一段和二段丙烯酸的总质量)的水溶液68.8g加入到55.2g丙烯酸(即,二段丙烯酸)中,然后加入0.011g抗坏血酸(0.02wt%,基于二段丙烯酸的质量)以得到二段丙烯酸水溶液(二段丙烯酸的质量浓度为44.4%)。此后,将步骤1得到的含有一段聚丙烯酸胶粒的悬浮液冷却至30℃,加入二段丙烯酸水溶液,氮气充分置换后,在350rpm的转速下搅拌30min,使得二段丙烯酸水溶液的丙烯酸液滴与一段聚丙烯酸胶粒彼此吸附并聚集。然后,边发生颗粒团聚边进行二段油包水反相悬浮聚合,得到含有二段聚丙烯酸胶粒的悬浮液。
3、酸碱中和
二段聚合结束后,将186.7g质量浓度为23%的氢氧化钠水溶液以2-3滴/秒的滴加速度滴入步骤2得到的含有二段聚丙烯酸胶粒的悬浮液。此时,加入的碱液与得到的二段聚丙烯酸胶粒发生酸碱中和反应,酸碱中和反应的温度为30℃,时间为10min,中和度70%,待体系温度恒定后,继续搅拌15min。
4、共沸脱水与表面交联
将步骤3得到的悬浮液升温至100℃进行共沸脱水并使得正庚烷回流,共脱除220g水(70%的水,基于四口圆底烧瓶中总共313g的水)。将体系温度调整为80℃,向悬浮液中加入质量浓度为1%的乙二醇二缩水甘油醚水溶液9.2g(乙二醇二缩水甘油醚用量为一段和二段丙烯酸总质量的0.08%),在此温度下反应1h进行表面交联。
5、溶剂脱除与干燥
将体系升温至120℃,蒸馏0.5h去除体系中剩余的水与正庚烷,此时可以观察到葡萄串状的高吸水性树脂(sap)粒子沉在烧瓶底部。最后,将其在100℃下干燥1.5h并筛分可得到所需粒径(150-710微米,占85%)的吸水性树脂产品,最终含水量为3%。
比较试验:
取本发明实施例1-6制备的吸水性树脂与竞品a、b和c进行基本性能比较,结果如表2所示。
表2吸水性树脂的基本性能比较
从表2可以看出,采用本发明(先聚合、后中和的反相悬浮聚合工艺)所得吸水性树脂与竞品a相比,尽管通液速率有所降低,但是1min纯水吸收量和可萃取物方面有所改善;与竞品b相比,本发明的吸水性树脂1min纯水吸收量和可萃取物方面大幅改善;而与竞品c相比,吸水性树脂尽管1min纯水吸收量降低和可萃取物增加,但是通液速率显著提高。
由于,吸水性树脂的各个性能是相互制约的,本发明制备的吸水性树脂,在其他性能降低程度可以接受(需要改善吸液倍率和离心保水性能)的情况下,获得了兼具有较好通液速率和1min纯水吸收量的sap颗粒,但是这为目前应用在超薄化复合型芯体市场的吸水性树脂的开发提供了新思路。本发明的这种独特的反相悬浮聚合工艺丰富了sap的制造方法,且为差异化性能sap的制备提供了一种可能。
- 还没有人留言评论。精彩留言会获得点赞!