一种基因点突变修复的新方法与流程
本发明属于基因修复技术领域,尤其是涉及一种基因点突变修复的新方法。
背景技术:
crispr/cas9系统是近年来发展起来的一种新的高效基因编辑技术,它可以在细胞水平对基因组进行删除、修改。crispr/cas系统自2013年初公布之后,很快便在全世界范围内得到了广泛地应用,并且被《science》杂志评为2015年十大科学突破之一。crispr/cas9是一种rna-蛋白质的复合体,常用的是cas9核酸酶,由1409个氨基酸组成,有2个重要的核酸酶结构域,分别是ruvc和hnh结构域。hnh结构域负责断裂通过靶向互补的dna单链,切割位点位于pam(protospacer-adjacentmotifs)序列上游3nt处。ruvc结构域负责断裂另一条dna链,切割位点位于pam序列上游3~8nt处。rna-dna双链在pam位点开始形成。cr-rna/trancr-rna结合在pam序列的附近,通过ruvc和hnh结构域诱导cas9核酸酶的活性。目前通过基因工程的方法已经将cr-rna和tracr-rna这两种rna,简化成具有相同功能的sgrna。
利用crispr/cas9系统对突变基因进行编辑在医学研究方面的应用颇为广泛,在hiv的治疗,遗传疾病治疗,如亨丁顿舞蹈病的治疗,癌症治疗,血液疾病方面以及眼部疾病等方面的治疗都取得了显著的进展,为疾病的研究打开了新的大门。
我们的前期研究发现了一种能导致女性原发性闭经的遗传突变(tingguoetal,2017,humanmolecμlargenetics),该突变位于msh5基因1459位点的g到c的突变,该突变导致msh5不能正常行驶功能,从而使得生殖细胞减数分裂过程不能完成,最终引起生殖细胞死亡。
技术实现要素:
有鉴于此,本发明旨在提出一种基因点突变修复的新方法,利用电转染技术和高效的基因编辑技术crispr/cas9系统相结合的治疗方法体外修复生殖细胞。
为达到上述目的,本发明的技术方案是这样实现的:
一种基因点突变修复的新方法,主要步骤如下:
(1)在目标基因组序列中,选定细胞基因突变位点为修复的基因位点;
(2)根据选定的基因修复位点,设计crispr/cas系统的sgrna;
(3)将sgrna构建到携带crispr/cas9蛋白的表达载体上;
(4)在突变位点的上游或下游附近选取一段正确的序列,并由此设计donordna,作为基因突变修复的模板;
(5)体外培养携带突变位点基因的细胞;
(6)利用电转染的方法,将sgrna和crispr/cas9的表达载体、donordna转染到细胞中。
进一步,选定基因的突变位点为msh5点。
进一步,该msh5点存在于女性生殖细胞中。
进一步,msh5突变位点所对应的sgrna序列为:
guide#1:ctatcgtagcgcccggaccaagg
guide#8:caaggagctgtacacgctgctgg。
进一步,guide#1的上游引物及下游引物序列分别为:
f-caccgctatcgtagcgcccggacca
r-aaactggtccgggcgctacgatagc;
guide#8的上游引物及下游引物序列分别为:
f-caccgcaaggagctgtacacgctgc
r-aaacgcagcgtgtacagctccttgc。
进一步,crispr/cas9蛋白的表达载体为pcs(egfp)载体。
进一步,donordna的序列为:
cagtttctctcagaggacaagctgcactatcgtagcgcccggaccaaggagctggacacgctgctgggagacctgcactgcgagatccggggtgaggagcccgtggtaggagggggcaggctgctctaac。
进一步,电转染的脉冲电源电压为33v,脉冲间隔50ms~950ms,正反向各4次。
需要说明的是,在临床上有许多生殖细胞发生异常都是由于基因突变导致的,本申请的方法是将cas9表达载体、sgrna,以及donordna与体外建立的携带msh5点突变的小鼠成纤维细胞系充分混合,然后对细胞系进行电转染处理,增加细胞的转染效率,并利用crispr/cas9系统对这种突变位点进行修复。通过这种处理使得cas9表达载体、sgrna和donordna进入生殖细胞中,从而将生殖细胞中的基因突变位点进行修复,并利用测序的方法检测突变位点修复的效率,从而为治疗女性原发性闭经疾病提供科学依据。
附图说明
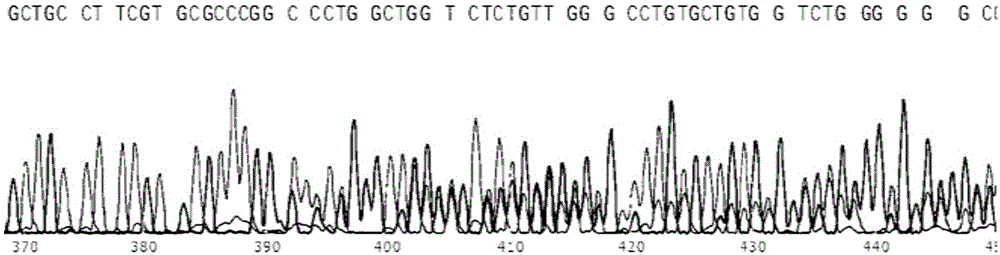
图1为extagpcr产物测序正向引物测序结果;
图2为extagpcr产物测序反向引物测序结果;
图3为t7ei酶切反应体系电泳检测结果。
具体实施方式
除有定义外,以下实施例中所用的技术术语具有与本发明所属领域技术人员普遍理解的相同含义。以下实施例中所用的试验试剂,如无特殊说明,均为常规生化试剂;所述实验方法,如无特殊说明,均为常规方法。
下面结合实施例来详细说明本发明。
以携带msh5点突变的小鼠为例,对msh5点突变修复的具体研究方案如下:
实施例一载体的构建
1、sgrna的设计:使用sgrna在线设计工具,在突变的位点附近选取合适的sgrna。
sgrna选择guide#1和guide#8,序列信息为:
guide#1ctatcgtagcgcccggaccaagg
guide#8caaggagctgtacacgctgctgg。
2、将sgrna构建到pcs(egfp)载体上,该表达载体携带cas9蛋白。
(1)设计引物:在上下游的引物中引入酶切位点bbsi,引物序列为:
guide#1:f-caccgctatcgtagcgcccggacca
r-aaactggtccgggcgctacgatagc
guide#8:f-caccgcaaggagctgtacacgctgc
r-aaacgcagcgtgtacagctccttgc
(2)引物退火成双链:引物用te溶解为200μm后进行退火
退火体系共20μl
混匀后,直接放入沸水中退火,直至冷却至室温,放于4℃。
(3)t4连接酶连接
pcs(egfp)载体用bbsi限制性内切酶37℃过夜酶切,并切胶回收获得线性化的载体,此载体用于连接;取1μl退火后的产物,加99μlddh2o稀释至100μl,稀释液用于连接。
连接体系共15μl
混匀后,16℃连接过夜。
(4)转化
取5μl连接产物于50μl感受态中,冰上静置25分钟,42℃热激30秒,冰上静置2分钟。加入450μl无抗的lb,37℃,200rpm,复苏1小时。
(5)涂板
取100μl复苏后的菌液涂于amp+的平板内,37℃培养过夜。
(6)挑取单克隆,用质粒小提试剂盒提取质粒,测序验证载体是否正确连接。
实施例二载体的切割效率检测
一、guide#1切割效率检测
将构建的载体用无内毒素质粒提取试剂盒提取质粒。
1、受精卵原核注射,进行囊胚鉴定,验证切割活性
将载体注射入原核期受精卵,待受精卵发育至囊胚期(4天左右),进行囊胚鉴定,检测是否进行了正确的切割。囊胚加入99μl裂解液,1μl蛋白酶k,56℃水浴过夜裂解。裂解后的产物于95℃处理5分钟,使蛋白酶k变性,产物可用于pcr。囊胚鉴定使用extag进行pcr扩增,将有目的条带的pcr产物送测序。
extagpcr体系50μl
extagpcr反应程序
pcr产物测序,测序结果具体见图1和图2,其中显示,在理论的切割位点开始出现套峰,说明有切割活性。
2、转染f9细胞,检测切割效率
用lipo3000转染24孔板的f9细胞。转染质粒的量为500ng,未转染组做对照。转染48小时后,收集细胞,加入裂解液,提取dna。提取dna之后,进行t7ei酶切和ta克隆,检测切割效率。
(1)t7ei酶切
①pcr扩增
扩增引物f:cccaagggatgaaaagccac
r:gggagagtaatgcggtctcg
②pcr产物纯化后,按照如下体系混合于ep管中:
③加热变性,退火复性处理
在1升烧杯中加入沸水,ep管放入沸水中,烧杯于室温冷却。
④t7ei酶切反应
上述反应体系中加入0.5μlt7ei酶,37℃,反应30分钟。立即加入2μldnaloadingbuffer(6x)。混匀后,65℃煮10分钟。2%琼脂糖电泳检测,电泳结果如图3所示。
图中pcs(egfp)代表转染空载,1、2、3分别代表转染pcs(egfp)+grna1#载体的三个平行孔。通过灰度分析,计算突变条带灰度/野生型条带灰度,得到最终的突变率约为12%。
(2)ta克隆:使用peasy-t1simplecloningkit
①pcr扩增
扩增引物f:cccaagggatgaaaagccac
r:gggagagtaatgcggtctcg
②pcr产物与peasy-t1simplecloningvector连接
连接体系
反应条件:将体系轻轻混合,于pcr中25℃反应10分钟
③转化
连接产物于50μl感受态中,冰上静置25分钟,42℃热激30秒,冰上静置2分钟。加入450μl无抗的lb,37℃,200rpm,复苏1小时。
④涂板
⑤阳性克隆检测
菌液pcr鉴定:用载体通用引物m13f和m13r进行pcr鉴定
阳性条带送测序:挑取60个单菌落,有阳性条带的是20个,测序结果显示,仅有3个在理论位点发生切割,效率约为15%。
实施例三设计donerdna
在突变位点的上游和下游附近选取一段正确的序列作为donor,送生工合成。序列为:
cagtttctctcagaggacaagctgcactatcgtagcgcccggaccaaggagctggacacgctgctgggagacctgcactgcgagatccggggtgaggagcccgtggtaggagggggcaggctgctctaac。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!