一种氧气调控的二氧化碳基嵌段共聚物的合成方法与流程
本发明属于高分子合成领域,具体地,涉及一种二氧化碳基嵌段共聚物的合成方法,更具体地涉及一种依靠氧气调控的二氧化碳基嵌段共聚物的合成。
背景技术:
传统高分子原料的日益枯竭,及其难降解特性带来的严峻环境污染使发展可再生、可降解高分子成为必然选择。基于二氧化碳和环氧化合物共聚合成的二氧化碳基聚碳酸酯具有优异的生物降解、生物相容及阻隔性能,是传统非降解高分子的理想替代品,在食品包装和农用地膜等领域极具应用前景。然而,脂肪族二氧化碳基聚碳酸酯分子链间作用力较弱,力学性能和热学稳定性较差,同时主链结构中缺少反应性的官能团,制约了二氧化碳基聚碳酸酯的应用拓展。
嵌段共聚物能够将多种聚合物的优良性能结合在一起,同时能够提高聚合物链段之间的相容性,因此通过引入共聚单体合成嵌段共聚物,是改善二氧化碳基聚碳酸酯物理和化学性能的理想方式。秦玉升等人利用稀土三元催化剂成功地制备得到环氧丙烷/二氧化碳/丙交酯的三元共聚物,其初始分解温度与聚碳酸亚丙酯相比提高了32℃,且其断裂伸长率达到了40.5%,结果表明丙交酯的加入有效地提高了聚碳酸亚丙酯的热力学性能。
目前二氧化碳基嵌段共聚物的合成的研究较少,合成方法以分步加料法和串联反应法为主。分步加料法合成过程中所用的催化剂和聚合方法保持不变,适用于多种环氧单体的嵌段共聚物的合成,但是需要在每种环氧单体反应结束后将进行产物分离。darensburg等人在反应体系中顺序加入环氧丙烷单体、环氧环己烷单体与二氧化碳发生共聚反应,以分步加料法合成二氧化碳基聚碳酸酯嵌段共聚物。而对于二氧化碳共聚反应来说,位阻效应和电子效应对环氧化物的反应活性影响较大,不同结构的环氧单体适应的催化剂和反应温度有较大差异,只有特定的催化剂和环氧单体的组合才能利用分步加料法合成二氧化碳基聚碳酸酯嵌段共聚物。串联法合成适用于非环氧单体合成二氧化碳基聚碳酸酯嵌段共聚物,吕小兵以活泼质子为链转移剂,合成末端为羟基的二氧化碳基聚碳酸酯二元醇,并利用二氧化碳基聚碳酸酯二元醇引发丙交酯的开环聚合反应合成了二氧化碳基聚碳酸酯嵌段共聚物。串联法合成二氧化碳基聚碳酸酯嵌段共聚物本质上是利用含羟基的大分子引发剂引发内酯的开环聚合反应,而适用开环反应的内酯种类较少,这限制了串联反应法的广泛应用。因此,分步加料法和串联反应法合成二氧化碳基嵌段共聚物都存在适用单体较少的弊端,相比较而言,一锅法合成更简单高效。
技术实现要素:
针对现有技术的以上缺陷或改进需求,本发明提供了一种氧气调控的二氧化碳基嵌段共聚物的合成方法,该合成方法选取金属钴中心催化体系,首先进行所述乙烯基单体的自由基聚合反应,然后通过氧气对催化中心的活化作用使所述环氧化物单体与二氧化碳进行阴离子开环共聚反应,最终得到分子量分布窄的二氧化碳基嵌段共聚物。该聚合方法以氧气作为外源刺激,便捷高效地实现了单金属中心串联催化自由基聚合和阴离子开环共聚反应,不仅在时间和空间上对聚合过程有效控制,并且对高分子聚合物微观结构实现了精确控制,更是对现有聚合机理转换体系的组分进一步优化,简化了聚合机理切换过程、降低成本,扩展了单体的选择范围,丰富了二氧化碳基嵌段共聚物的结构,由此解决现有技术采用分步加料法和串联反应法制备二氧化碳基嵌段共聚物步骤复杂、产物需要分离,以及适用单体少、具有应用局限性的技术问题。
为实现上述目的,按照本发明的一个方面,提供了一种二氧化碳基嵌段共聚物的合成方法,以金属有机钴配合物为催化剂,以乙烯基单体和环氧化物单体为原料,包括如下步骤:
乙烯基单体在金属有机钴配合物的调控下进行自由基聚合反应,通过钴-碳键的可逆均裂建立co(ii)和co(iii)的平衡,形成可控自由基聚合体系;
通过氧气对金属有机钴配合物进行活化作用,在所述自由基聚合体系中发生钴-碳键的氧原子插入反应,形成钴-氧键作为阴离子聚合催化位点;
环氧化物单体与二氧化碳在所述阴离子聚合催化位点进行阴离子开环共聚反应,得到二氧化碳基嵌段共聚物。
优选地,所述的合成方法,包括如下步骤:
s1:在无水且隔绝氧气的条件下,将乙烯基单体、环氧化物单体、金属有机钴配合物、自由基引发剂以及有机碱在反应容器中混合均匀形成预反应混合液;其中,所述乙烯基单体与所述金属有机钴配合物的摩尔比为100:1~1000:1;所述乙烯基单体与所述自由基引发剂的摩尔比为50:1~250:1;所述环氧化物单体与所述金属有机钴配合物的摩尔比为1000:1~2500:1;所述有机碱与所述金属有机钴配合物的摩尔比为0.5:1~5:1;
s2:将步骤s1得到的预反应混合液置于预定反应温度,所述乙烯基单体在自由基引发剂引发、金属有机钴配合物调控作用下进行自由基聚合反应;
s3:步骤s2所述自由基聚合反应结束后,向所述反应容器中充入预定压力的氧气,并将所述反应容器内的温度控制在预定温度,将金属有机钴配合物活化成阴离子开环共聚催化体系;
s4:将步骤s3所述反应容器中的氧气释放,充入预定压力的二氧化碳,控制反应温度在室温(20~30℃),并记录反应时间,步骤s1所述环氧化物单体与二氧化碳进行阴离子开环共聚反应;
s5:反应一段时间后,释放反应容器内的二氧化碳,结束步骤s4所述阴离子开环共聚反应,除去未反应的原料,得到分子量分布指数为1.11~1.35的二氧化碳基嵌段共聚物。具体反应时间长度依据目标嵌段共聚物的聚合程度需求而定。
进一步优选地,所述乙烯基单体为醋酸乙烯酯、丙烯酸甲酯、丙烯酸正丁酯、丙烯酸叔丁酯、丙烯腈、n-乙烯基吡咯烷酮、n,n-二甲基丙烯酰胺、n,n-二乙基丙烯酰胺和n-丙烯酰吗啉中的至少一种;
所述环氧化物单体为环氧乙烷、环氧丙烷、环氧氯丙烷、1,2-环氧丁烷、环氧环戊烷、环氧环己烷和苯乙烯环氧烷烃中的至少一种;
所述金属有机钴配合物为具有以下结构的钴系双希夫碱配合物:
式中,r1为含取代基的烷基链,所述烷基链为c1-c20的烷基或c1-c20的烷氧基,所述取代基为卤素、硝基、氨基或氰基;
r2,r3和r4各自独立的选自h、c1-c20的烷基、c1-c20的烷氧基、c6-c20的芳基、含有取代基团的c1-c20烷基和含有取代基团的c6-c20芳基;
其中,r2,r3和r4各自独立的选自h、c1-c20的烷基、c6-c20的芳基、含有取代基团的c1-c20烷基和含有取代基团的c6-c20芳基时,r2与r3之间或r3和r4之间可各自独立形成封闭环。
r2,r3和r4各自独立的选自h、c1-c20的烷基、c1-c20的烷氧基和c1-c20氟取代的烷基时,各取代基之间不成环。
所述自由基引发剂为偶氮二异丁腈、偶氮二异庚腈、过氧化二苯甲酰、过氧化十二酰和过氧化碳酸二环己酯引发剂中的至少一种;
所述有机碱为1,8-二氮杂二环十一碳-7-烯、1,5,7-三氮杂二环[4.4.0]癸-5-烯、7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯、7-乙基-1,5,7-三氮杂二环[4.4.0]癸-5-烯、7-叔丁基-1,5,7-三氮杂二环[4.4.0]癸-5-烯和4-二甲氨基吡啶中的任意一种。
优选地,步骤s1所述预反应混合液是在保护性气体氛围下搅拌混合10~30分钟得到的。
进一步优选地,所述保护性气体为氮气或者惰性气体。
优选地,步骤s2所述预定反应温度为50℃~100℃。
优选地,步骤s2所述自由基聚合反应时间不少于2个小时。
进一步优选地,所述自由基聚合反应时间为2~24小时。
优选地,所述自由基聚合反应是通过冷却反应体系结束的。
优选地,步骤s3所述预定温度为50℃~100℃。
优选地,步骤s3所述氧气的预定压力不低于1mpa。
进一步优选地,所述氧气的预定压力为1mpa~6mpa。
优选地,步骤s4所述二氧化碳的预定压力不低于1mpa。
进一步优选地,所述二氧化碳的预定压力为1mpa~5mpa。
优选地,步骤s5所述除去未反应的原料具体为:通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱。
总体而言,通过本发明所构思的以上技术方案与现有技术相比,能够取得下列有益效果:
(1)针对现阶段单金属活性中心串联催化不同聚合反应机理的挑战,本发明提供的氧气调控的二氧化碳基嵌段共聚物的合成方法,利用氧气为外界刺激源调控钴-碳键向钴-氧键的过渡,金属钴活性中心串联催化自由基聚合与阴离子开环共聚,最终合成分子量分布窄的二氧化碳基嵌段共聚物。
具体来说,首先金属有机钴配合物调控的自由基聚合过程中钴-碳键的可逆均裂建立co(ii)和co(iii)的平衡,同时在预定的加热条件下促进该平衡动态进行,形成高效、适用性强的可控自由基聚合体系,聚合机理如下:
接着在聚合结束后,通入预定压力氧气即开始钴-碳键的氧原子插入反应,得到的钴-氧键为阴离子聚合催化位点,催化位点活化过程如下:
随后,释放反应容器中的氧气,充入预定压力二氧化碳,所述环氧单体即开始与二氧化碳开始阴离子开环共聚反应,最终得到分子量分布窄的二氧化碳基嵌段共聚物。
氧气调控的二氧化碳基嵌段共聚物的合成方法涉及自由基聚合和阴离子开环共聚机理的转换,聚合方法过程如下:
(2)目前高分子合成领域中,将单体精确地插入到聚合物链中形成序列可控聚合物仍是一个巨大挑战,且低活性乙烯基单体产生的自由基活性非常高,易发生自由基转移和中止等副反应。本发明提供的金属有机钴配合物调控的自由基聚合反应可实现对低活性乙烯基单体的可控聚合,确保聚合物微观链结构和分子链长度的精密调控,同时阴离子开环共聚反应的环氧化物单体选择性广、可控性强,最终可根据实际需要对嵌段聚合物进行设计合成。
(3)针对现阶段二氧化碳基嵌段共聚物的合成,本发明创造性地在金属有机钴配合物调控的自由基聚合基础上,通过氧气对催化位点的活化使环氧化物单体与二氧化碳进行阴离子开环共聚反应,最终得到分子量分布窄的二氧化碳基嵌段共聚物。相较于分步加料法省略了分步合成的中间产物纯化过程和产物后修饰过程,规避了传统的分步法带来的繁琐操作,同时自由基聚合适用单体、范围广泛,能极大地丰富二氧化碳基嵌段共聚物的结构,克服串联法单体适用范围小的缺点,本发明合成方法为二氧化碳基嵌段共聚物的性能定向设计提供了基础。
附图说明
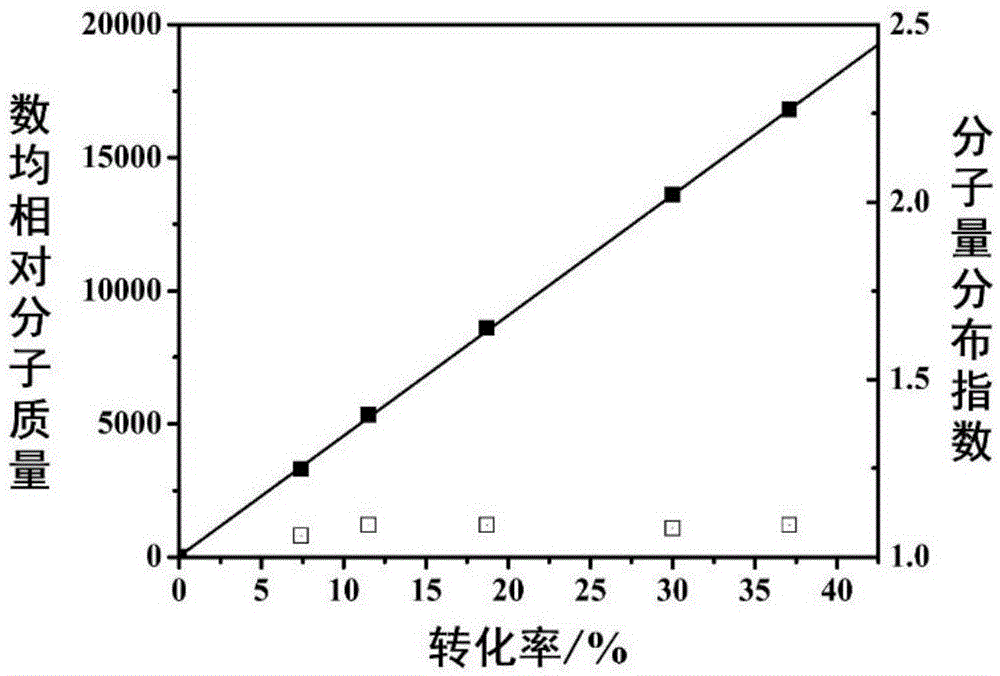
图1为实施例3中聚合体系[醋酸乙烯酯]0:[环氧丙烷]0:[n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物]0:[偶氮二异丁腈]0:[7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯]0=500:2000:1:7:1在60℃反应得到聚醋酸乙烯酯的分子量和分子量分布曲线;
图2为实施例6中聚合体系[醋酸乙烯酯]0:[环氧丙烷]0:[n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物]0:[偶氮二异丁腈]0:[7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯]0=500:2000:1:7:1在60℃反应得到聚醋酸乙烯酯的聚合动力学曲线;
图3为实施例8中聚合体系[醋酸乙烯酯]0:[环氧丙烷]0:[n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物]0:[偶氮二异丁腈]0:[7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯]0=500:2000:1:7:1反应得到的二氧化碳基嵌段共聚物核磁共振氢谱(1hnmr,cdcl3)图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
本发明提供了一种氧气调控的二氧化碳基嵌段共聚物的合成方法,该合成方法以金属有机钴配合物为催化体系,首先乙烯基单体在金属有机钴配合物调控下进行自由基聚合反应,然后通过氧气对金属有机钴配合物进行活化作用使环氧化物单体与二氧化碳进行阴离子开环共聚反应,得到分子量分布窄的二氧化碳基嵌段共聚物,聚合体系包括乙烯基单体、环氧化物单体、金属有机钴配合物、自由基引发剂以及有机碱,该聚合方法以氧气作为外源刺激,便捷地实现了单金属中心串联催化自由基聚合和阴离子开环共聚机理,对高分子聚合物微观结构实现了精确控制,规避了传统的分步法带来的繁琐过程,扩展了单体的选择范围,丰富了二氧化碳基嵌段共聚物的结构。
以下为实施例:
实施例1:
在无水且隔绝氧气的手套箱中称取6.4克丙烯酸正丁酯、8.8克环氧乙烷、60.3毫克n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物、114.9毫克偶氮二异丁腈以及15.3毫克7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯加入到高压反应釜中,磁力搅拌10分钟后,得到预反应混合液;
将得到的预反应混合液加热到60℃,且在60℃恒定温度下,醋酸乙烯酯在偶氮二异丁腈引发、n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物调控作用下进行自由基聚合反应;
自由基聚合反应6小时后,向高压反应釜中充入2mpa的氧气,将高压反应釜内的温度控制在60℃,金属有机钴配合物活化成阴离子开环共聚催化体系;
活化反应12小时后,将高压反应釜内的氧气释放,然后充入3mpa的二氧化碳,控制高压反应釜的温度处于室温,环氧丙烷单体与二氧化碳进行阴离子开环共聚反应;
反应24小时后,缓慢释放高压反应釜内的二氧化碳,结束阴离子开环共聚反应。用5~8毫升二氯甲烷将产物溶解,通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱,干燥得到二氧化碳基嵌段共聚物,其分子量分布指数为1.21。
实施例2:
在无水且隔绝氧气的手套箱中称取6.4克丙烯酸叔丁酯、14.5克环氧丙烷、60.3毫克n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物、114.9毫克偶氮二异丁腈以及15.3毫克7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯加入到高压反应釜中,磁力搅拌10分钟后,得到预反应混合液;
将得到的预反应混合液加热到60℃,且在60℃恒定温度下,醋酸乙烯酯在偶氮二异丁腈引发、n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物调控作用下进行自由基聚合反应;
自由基聚合反应7小时后,向高压反应釜中充入1mpa的氧气,将高压反应釜内的温度控制在60℃,金属有机钴配合物活化成阴离子开环共聚催化体系;
活化反应12小时后,将高压反应釜内的氧气释放,然后充入2mpa的二氧化碳,控制高压反应釜的温度处于室温,环氧丙烷单体与二氧化碳进行阴离子开环共聚反应;
反应24小时后,缓慢释放高压反应釜内的二氧化碳,结束阴离子开环共聚反应。用5~8毫升二氯甲烷将产物溶解,通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱,干燥得到二氧化碳基嵌段共聚物,其分子量分布指数为1.11。
实施例3:
在无水且隔绝氧气的手套箱中称取4.3克醋酸乙烯酯、12克氧化苯乙烯、59.7毫克n,n-双(3,5-二叔丁基亚水杨基-1,2-邻苯二胺)钴配合物、173.8毫克偶氮二异庚腈以及15.3毫克7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯加入到高压反应釜中,磁力搅拌10分钟后,得到预反应混合液;
将得到的预反应混合液加热到60℃,且在60℃恒定温度下,醋酸乙烯酯在偶氮二异庚腈引发、n,n-双(3,5-二叔丁基亚水杨基-1,2-邻苯二胺)钴配合物调控作用下进行自由基聚合反应;
自由基聚合反应7小时后,向高压反应釜中充入3mpa的氧气,将高压反应釜内的温度控制在60℃,金属有机钴配合物活化成阴离子开环共聚催化体系;
活化反应12小时后,将高压反应釜内的氧气释放,然后充入4mpa的二氧化碳,控制高压反应釜的温度处于室温,环氧丙烷单体与二氧化碳进行阴离子开环共聚反应;
反应24小时后,缓慢释放高压反应釜内的二氧化碳,结束阴离子开环共聚反应。用5~8毫升二氯甲烷将产物溶解,通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱,干燥得到二氧化碳基嵌段共聚物,其分子量分布指数为1.15。
图1为本实施例中聚合体系[醋酸乙烯酯]0:[环氧丙烷]0:[n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物]0:[偶氮二异丁腈]0:[7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯]0=500:2000:1:7:1在60℃反应得到聚醋酸乙烯酯的分子量和分子量分布曲线;从图1可以看出可控自由基聚合体系中,数均相对分子质量随着转化率的提高而不断增大,理论数均相对分子质量与实际数均相对分子质量吻合,且分子量分布指数均低于1.11。
实施例4:
在无水且隔绝氧气的手套箱中称取5.3克丙烯腈、11.6克环氧丙烷、60.3毫克n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物、114.9毫克偶氮二异丁腈以及16.7毫克7-乙基-1,5,7-三氮杂二环[4.4.0]癸-5-烯加入到高压反应釜中,磁力搅拌10分钟后,得到预反应混合液;
将得到的预反应混合液加热到60℃,且在60℃恒定温度下,醋酸乙烯酯在偶氮二异丁腈引发、n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物调控作用下进行自由基聚合反应;
自由基聚合反应6小时后,向高压反应釜中充入4mpa的氧气,将高压反应釜内的温度控制在60℃,金属有机钴配合物活化成阴离子开环共聚催化体系;
活化反应12小时后,将高压反应釜内的氧气释放,然后充入4mpa的二氧化碳,控制高压反应釜的温度处于室温,环氧丙烷单体与二氧化碳进行阴离子开环共聚反应;
反应24小时后,缓慢释放高压反应釜内的二氧化碳,结束阴离子开环共聚反应。用5~8毫升二氯甲烷将产物溶解,通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱,干燥得到二氧化碳基嵌段共聚物,其分子量分布指数为1.20。
实施例5:
在无水且隔绝氧气的手套箱中称取0.9克醋酸乙烯酯、14.4克1,2-环氧丁烷、54.9毫克n,n-双(3,5-二叔丁基亚水杨基-1,2-乙二胺)钴配合物、169.59毫克过氧化二甲苯酰以及15.2毫克1,8-二氮杂二环十一碳-7-烯加入到高压反应釜中,磁力搅拌10分钟后,得到预反应混合液;
将得到的预反应混合液加热到60℃,且在60℃恒定温度下,醋酸乙烯酯在过氧化二甲苯酰、n,n-双(3,5-二叔丁基亚水杨基-1,2-乙二胺)钴配合物调控作用下进行自由基聚合反应;
自由基聚合反应8小时后,向高压反应釜中充入4mpa的氧气,将高压反应釜内的温度控制在60℃,金属有机钴配合物活化成阴离子开环共聚催化体系;
活化反应24小时后,将高压反应釜内的氧气释放,然后充入2mpa的二氧化碳,控制高压反应釜的温度处于室温,环氧丙烷单体与二氧化碳进行阴离子开环共聚反应;
反应48小时后,缓慢释放高压反应釜内的二氧化碳,结束阴离子开环共聚反应。用5~8毫升二氯甲烷将产物溶解,通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱,干燥得到二氧化碳基嵌段共聚物,其分子量分布指数为1.14。
实施例6:
在无水且隔绝氧气的手套箱中称取4.3克醋酸乙烯酯、19.6克环氧环己烷、60.3毫克n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物、114.9毫克偶氮二异丁腈以及15.3毫克7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯加入到高压反应釜中,磁力搅拌10分钟后,得到预反应混合液;
将得到的预反应混合液加热到60℃,且在60℃恒定温度下,醋酸乙烯酯在偶氮二异丁腈引发、n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物调控作用下进行自由基聚合反应;
自由基聚合反应7小时后,向高压反应釜中充入2mpa的氧气,将高压反应釜内的温度控制在60℃,金属有机钴配合物活化成阴离子开环共聚催化体系;
活化反应12小时后,将高压反应釜内的氧气释放,然后充入4mpa的二氧化碳,控制高压反应釜的温度处于室温,环氧丙烷单体与二氧化碳进行阴离子开环共聚反应;
反应48小时后,缓慢释放高压反应釜内的二氧化碳,结束阴离子开环共聚反应。用5~8毫升二氯甲烷将产物溶解,通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱,干燥得到二氧化碳基嵌段共聚物,其分子量分布指数为1.22。
图2为本实施例中聚合体系[醋酸乙烯酯]0:[环氧丙烷]0:[n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物]0:[偶氮二异丁腈]0:[7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯]0=500:2000:1:7:1在60℃反应得到聚醋酸乙烯酯的聚合动力学曲线;从图2可以反应动力学均呈现一级动力学行为,表明聚合过程中链增长自由基的浓度处于稳态。
实施例7:
在无水且隔绝氧气的手套箱中称取5.5克醋酸乙烯酯、16.8克环氧环己烷、59.7毫克n,n-双(3,5-二叔丁基亚水杨基-1,2-邻苯二胺)钴配合物、79.7毫克过氧化十二酰以及13.9毫克1,5,7-三氮杂二环[4.4.0]癸-5-烯加入到高压反应釜中,磁力搅拌10分钟后,得到预反应混合液;
将得到的预反应混合液加热到60℃,且在60℃恒定温度下,醋酸乙烯酯在过氧化十二酰、n,n-双(3,5-二叔丁基亚水杨基-1,2-邻苯二胺)钴配合物调控作用下进行自由基聚合反应;
自由基聚合反应6小时后,向高压反应釜中充入2mpa的氧气,将高压反应釜内的温度控制在60℃,金属有机钴配合物活化成阴离子开环共聚催化体系;
活化反应12小时后,将高压反应釜内的氧气释放,然后充入3mpa的二氧化碳,控制高压反应釜的温度处于室温,环氧丙烷单体与二氧化碳进行阴离子开环共聚反应;
反应24小时后,缓慢释放高压反应釜内的二氧化碳,结束阴离子开环共聚反应。用5~8毫升二氯甲烷将产物溶解,通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱,干燥得到二氧化碳基嵌段共聚物,其分子量分布指数为1.25。
实施例8:
在无水且隔绝氧气的手套箱中称取4.3克醋酸乙烯酯、11.6克环氧丙烷、54.9毫克n,n-双(3,5-二叔丁基亚水杨基-1,2-乙二胺)钴配合物、114.9毫克偶氮二异丁腈以及15.3毫克7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯加入到高压反应釜中,磁力搅拌10分钟后,得到预反应混合液;
将得到的预反应混合液加热到60℃,且在60℃恒定温度下,醋酸乙烯酯在偶氮二异丁腈引发、n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物调控作用下进行自由基聚合反应;
自由基聚合反应6小时后,向高压反应釜中充入3mpa的氧气,将高压反应釜内的温度控制在60℃,金属有机钴配合物活化成阴离子开环共聚催化体系;
活化反应24小时后,将高压反应釜内的氧气释放,然后充入4mpa的二氧化碳,控制高压反应釜的温度处于室温,环氧丙烷单体与二氧化碳进行阴离子开环共聚反应;
反应48小时后,缓慢释放高压反应釜内的二氧化碳,结束阴离子开环共聚反应。用5~8毫升二氯甲烷将产物溶解,通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱,干燥得到二氧化碳基嵌段共聚物,其分子量分布指数为1.12。
图3为本实施例中聚合体系[醋酸乙烯酯]0:[环氧丙烷]0:[n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物]0:[偶氮二异丁腈]0:[7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯]0=500:2000:1:7:1反应得到的二氧化碳基嵌段共聚物核磁共振氢谱(1hnmr,cdcl3)图。从该图中可以得出特征峰f(4.2ppm)为聚碳酸亚丙酯的亚甲基的氢位移,特征峰d(5.0ppm)为聚碳酸亚丙酯的次甲基的氢位移,特征峰b(4.9ppm)为聚醋酸乙烯酯的次甲基的氢位移,通过对聚合物的核磁分析表明得到目标二氧化碳基嵌段共聚物。
实施例9:
在无水且隔绝氧气的手套箱中称取5.0克n,n-二甲基丙烯酰胺、18.5克环氧氯丙烷、60.3毫克n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物、114.9毫克偶氮二异丁腈以及15.3毫克7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯加入到高压反应釜中,磁力搅拌10分钟后,得到预反应混合液;
将得到的预反应混合液加热到60℃,且在60℃恒定温度下,醋酸乙烯酯在偶氮二异丁腈引发、n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物调控作用下进行自由基聚合反应;
自由基聚合反应7小时后,向高压反应釜中充入4mpa的氧气,将高压反应釜内的温度控制在60℃,金属有机钴配合物活化成阴离子开环共聚催化体系;
活化反应24小时后,将高压反应釜内的氧气释放,然后充入4mpa的二氧化碳,控制高压反应釜的温度处于室温,环氧丙烷单体与二氧化碳进行阴离子开环共聚反应;
反应24小时后,缓慢释放高压反应釜内的二氧化碳,结束阴离子开环共聚反应。用5~8毫升二氯甲烷将产物溶解,通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱,干燥得到二氧化碳基嵌段共聚物,其分子量分布指数为1.35。
实施例10:
在无水且隔绝氧气的手套箱中称取7.1克n-丙烯酰吗啉、11.6克环氧丙烷、60.3毫克n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物、169.5毫克偶氮二异丁腈以及7.6毫克7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯加入到高压反应釜中,磁力搅拌10分钟后,得到预反应混合液;
将得到的预反应混合液加热到60℃,且在60℃恒定温度下,醋酸乙烯酯在偶氮二异丁腈引发、n,n-双(3,5-二叔丁基亚水杨基-1,2-环己二胺)钴配合物调控作用下进行自由基聚合反应;
自由基聚合反应6小时后,向高压反应釜中充入2mpa的氧气,将高压反应釜内的温度控制在60℃,金属有机钴配合物活化成阴离子开环共聚催化体系;
活化反应24小时后,将高压反应釜内的氧气释放,然后充入4mpa的二氧化碳,控制高压反应釜的温度处于室温,环氧丙烷单体与二氧化碳进行阴离子开环共聚反应;
反应24小时后,缓慢释放高压反应釜内的二氧化碳,结束阴离子开环共聚反应。用5~8毫升二氯甲烷将产物溶解,通过200~300目中性氧化铝除掉催化剂和引发剂,在正己烷中沉淀除去未反应的单体和有机碱,干燥得到二氧化碳基嵌段共聚物,其分子量分布指数为1.30。
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!