具有6.3T矫顽力的钴-萘环氮氧自由基分子磁体材料及其制备方法与流程
本发明属于功能材料制备技术领域,涉及一种具有6.3t矫顽力钴-氮氧自由基磁体及其制备方法。
背景技术:
分子磁体由于在低温下可以展现磁弛豫和/或磁滞回线,是一种重要的潜在磁性存储材料,也因此受到科学家的广泛关注。鉴于分子磁体在高密度存储领域表现出来的巨大潜力,科学家致力于研究如何早日实现分子磁体的实际应用。虽然已有大量性能优异的分子磁体见诸报道,然而大部分的分子磁体性质仍然无法与传统磁体相比,因此难以满足实际的应用。合成具有高阻塞温度、高有效能垒和高矫顽场的分子磁体一直是这个领域最主要的挑战和目标。其中,很多实际应用都要求磁体具有大的矫顽场,例如在磁存储中,大的矫顽场可以有效减小退磁场,从而大大提高记录信号的稳定性,降低数据信息位损坏的危险。在过去几年间,陆续有一些具有大的矫顽场的分子磁体被报道,但是这些分子多是基于金属有机的体系,所以合成难度大,实验条件严苛,样品表征困难,较难应用于实际。
技术实现要素:
本发明的目的是:针对目前构筑具有大矫顽场的分子磁体的方法所呈现出的一些弊端,如操作复杂、样品稳定性差、表征困难等,提供一种用稳定的萘环氮氧自由基作为配体构筑高矫顽场的分子磁体及其制备方法。
氮氧自由基(nits)是一类易于合成、结构稳定且可修饰的自由基配体,相比于一般的抗磁性配体,自由基本身作为一种自旋载体,桥联过渡或稀土金属体系更容易产生强的磁耦合作用,从而得到具有高的有效能垒和大的矫顽场的分子磁体。同时相比于已报道的金属有机体系,在同样可获得大的矫顽场的分子基磁体的前提下,氮氧自由基体系具有操作简单、样品稳定性高、表征易于进行等明显优势。
为此,本发明选用萘环氮氧自由基为桥联配体,合成了基于链结构的分子磁体,该材料是由六氟乙酰丙酮钴与氮氧自由基组装成的一维配合物,在80℃下搅拌5分钟,室温冷却后过滤,滤液在室温下静置三天,得到棒状的黑色晶体。本发明方法操作简单,产物的产率、纯度和稳定性很高,是构筑具有大的矫顽场的分子磁体的一种有效途径。
本发明的技术方案:
一种钴-萘环氮氧自由基分子磁体材料,其化学式为:[co(hfac)2(etonapnit)]n,其中n为1到正无穷的自然数,hfac为六氟乙酰丙酮,etonapnit为-(2-乙氧基-1-萘基)-4,4,5,5-四甲基咪唑啉-3-氧化-1-氧基自由基;晶体结晶在正交晶系,空间群为p212121,晶胞参数为:
一种钴-萘环氮氧自由基分子磁体材料的制备方法,包括以下步骤:
1)参考文献方法合成自由基etonapnit(ullmanprocedure);
2)将co(hfac)2·2h2o的正己烷溶液在90-100℃下搅拌回流2小时以上,缓慢降温后加入etonapnit的二氯甲烷溶液,反应5分钟以上,然后降至室温并过滤;
3)将上述滤液置于容器中,用薄膜封口,并置于干燥器中室温挥发,三天后即得到目标产物。
步骤2)所述co(hfac)2·2h2o的正已烷溶液的浓度为0.0013mol/l。所述etonapnit的二氯甲烷溶液的浓度为0.008mol/l。所述co(hfac)2·2h2o与etonapnit的摩尔比为1:1。
【本发明的优点和有益效果】
该配合物的制备方法简单,反应条件温和,产率较高,且具有很好的空气稳定性。该配合物在零场下展现出慢磁驰豫行为,且在2k时具有非常大的磁滞回环,矫顽场接近6.3t。这种具有大的矫顽场的链状分子磁体材料,可以有效减少信息存储器件在环境微扰下产生的信息丢失情况,因此在高密度信息存储领域具有非常高的潜在应用价值。
【附图说明】
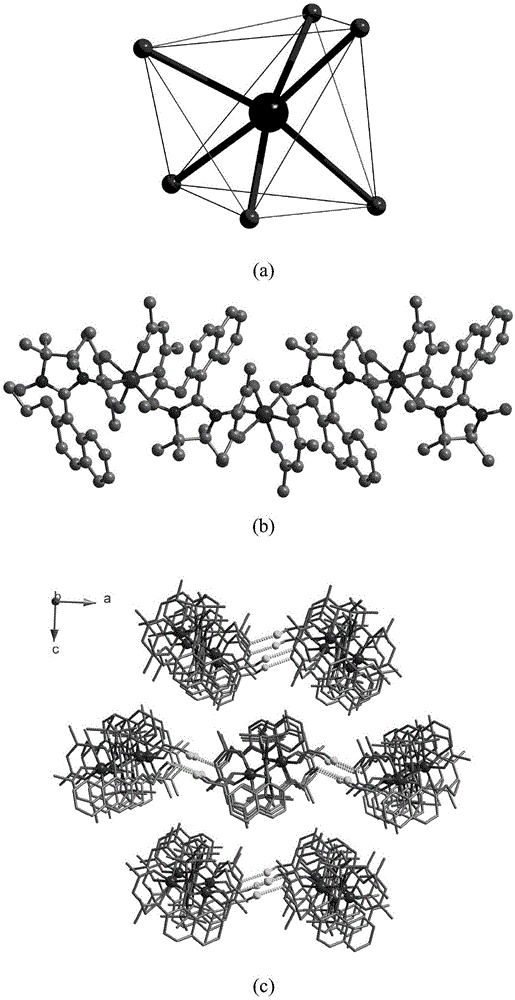
图1为该钴-萘环氮氧自由基配合物材料的单晶衍射结构图,其中:(a)为该材料中co2+离子的配位模式图;(b)为该材料的一维链状结构示意图;(c)为该化合物从b方向观察到的三维堆积图。
图2为该材料的粉末x-射线衍射图。
图3为该材料的静态磁学性质测试谱图,其中,(a)为该材料的在2-300k的直流磁化率测试图;(b)为该材料在2k下的磁滞回线。
图4为该材料的动态磁性质表征谱图,其中,(a)为该材料在零场下的交流扫温磁化率的虚部曲线;(b)为该材料在零场下的交流扫频磁化率的虚部曲线;(c)为该材料的零场下的cole-cole图。
【具体实施方式】
实施例1:
所述的钴-萘环氮氧自由基分子磁体材料的制备方法,包括以下步骤:
1)etonapnit的合成:
将0.74g(5mmol)的2,3-二甲基-2,3-二羟胺基丁烷加入25ml圆底烧瓶中,然后加入10ml甲醇和10ml二氯甲烷的混合溶剂溶解,搅拌10分钟后加入1.00g(5mmol)的2-乙氧基-1-萘甲醛,在氩气保护下45℃搅拌两天,得到浅黄色油状液体,减压蒸去溶剂后溶于80ml三氯甲烷中,冰浴下剧烈搅拌10分钟,向其中加入30ml含有1.1g高碘酸钠的水溶液,继续冰浴搅拌约三十分钟,随后进行分液、洗涤水相、干燥有机相、减压蒸馏,最终得到深紫色油状物;选用石油醚与乙酸乙酯体积比为5:1的混合溶剂作为淋洗剂,对油状物进行色谱柱分离,收集紫色色带,减压蒸馏后得到深紫色的微晶,即为etonapnit,产率35%。
2)[co(hfac)2(etonapnit)]n的合成:
将co(hfac)2·2h2o加入30ml正己烷中形成悬浮液,在90℃下搅拌回流超过两小时后降至80℃,加入etonapnit的二氯甲烷溶液,反应10分钟后,自然冷却至室温过滤,将滤液置于容器中,用薄膜封口,并置于干燥器中室温挥发,约三天后得到黑色棒状晶体,产率70%。
其中,co(hfac)2·2h2o的浓度为0.0013mol/l。etonapnit的浓度为0.008mol/l。co(hfac)2·2h2o与etonapnit的摩尔比为1:1。
实施例2:
所述的钴-萘环氮氧自由基分子磁体材料的制备方法,包括以下步骤:
1)etonapnit的合成:
将0.45g(3mmol)的2,3-二甲基-2,3-二羟胺基丁烷加入25ml圆底烧瓶中,然后加入8ml甲醇和5ml二氯甲烷的混合溶剂溶解,搅拌5分钟后加入0.65g(3.2mmol)的2-乙氧基-1-萘甲醛,在氩气保护下30℃搅拌36小时,得到浅黄色油状液体,溶于50ml的二氯甲烷中,冰浴下搅拌5分钟,向其中加入30ml含0.65g高碘酸钠的水溶液,继续冰浴反应25分钟,随后进行分液、洗涤水相、干燥有机相、减压蒸馏,最终得到深紫色油状物质;用石油醚与乙酸乙酯体积比为3:1的溶剂作为淋洗剂,对油状物进行色谱柱分离,收集紫色色带,减压蒸馏后得到深紫色的微晶,即为etonapnit,产率32%。
2)[co(hfac)2(etonapnit)]n的合成:
将co(hfac)2·2h2o加入25ml正己烷中形成悬浮液,在100℃下搅拌回流超过两小时后降至75℃,加入etonapnit的二氯甲烷溶液,反应5分钟后,自然冷却至室温过滤,将滤液置于容器中,用薄膜封口,并置于干燥器中室温挥发,约三天后得到黑色棒状晶体,产率65%。
其中,co(hfac)2·2h2o的浓度为0.0016mol/l。etonapnit的浓度为0.008mol/l。co(hfac)2·2h2o与etonapnit的摩尔比为1:1。
该钴-萘环氮氧自由基分子磁体的结构测定:
配合物的单晶结构测定在oxfordsupernova型x-射线单晶衍射仪上完成,使用经石墨单色器单色化的mo-kα射线
表1配合物的晶体学数据
该钴-萘环氮氧自由基配合物的磁性测试:
磁性表征在quantumdesignmpmsvsmsquid上进行;对所有实验数据采用pascal’s常数进行抗磁校正。具体测试方法为:取25mg粉末样品装入磁性测试胶囊,然后对其进行静态和动态磁学性质的测试研究。
图3(a)是对该钴-萘环氮氧自由基配合物进行直流磁化率测试,300k的χmt值为4.14cm3mol-1k,大于一个仅考虑自旋贡献的coii加上一个s=1/2自由基的理论值2.25cm3mol-1k,归因于高自旋的coii离子会产生大的轨道贡献。图3(b)为配合物在2k时的磁滞回线。图4(a)和图4(b)分别是对该配合物在零外加场下的扫温和扫频交流磁化率曲线,他们均显示明显的温度和频率依赖性,说明配合物在零场下具有慢磁驰豫行为。图4(c)是配合物的扫频磁化率的虚部对实部数据作图得到的cole-cole图,它呈现出两个部分重叠的近半圆形状,用两个线性叠加的debye模型对数据进行拟合。
- 还没有人留言评论。精彩留言会获得点赞!