一种基因编辑方法在东方拟无枝酸菌中的应用与流程
本发明属于基因工程技术领域,具体地说,是关于一种基因编辑方法在东方拟无枝酸菌中的应用。
背景技术:
crispr/cas9系统,这是目前开发的一种快速、简单的对基因组进行定点编辑的新技术,只需要将一个单一的cas9蛋白与sgrna相结合,就能识别并切割特定dna序列完成高效的基因编辑。利用这一特点,cas9系统可成功与体外重组系统进行联用,直接从染色体上精确克隆片段dna。自2013年crispr/cas技术应用于基因敲除以后,这种构建快速、结构简单,高效的分子工具很快便被大家所接受,如今已成功应用于包括水稻、小麦、疟原虫、果蝇、斑马鱼以及哺乳动物等真核生物中。不仅在真核生物中,此系统在原核生物,如大肠杆菌以及微生物药物重要来源的链霉菌等体内基因功能研究方面也显示出优于传统的同源重组等其它现有技术之处。cas9系统也被证明可以成功与体内重组系统联用进行大片段dna的精确克隆。
万古霉素是众多耐药菌(如甲氧西林耐药的金黄葡萄球菌)引起感染的首选药物。东方拟无枝酸菌a.orientalis为万古霉素产生菌,对该菌应用基因工程技术进行基因编辑,对于提高万古霉素的产量、了解万古霉素的生物合成机制以及挖掘万古霉素衍生物具有重要意义。然而,目前传统的同源重组方法进行遗传操作,效率相对较低。于是本发明尝试将crispr/cas9基因编辑系统引入东方拟无枝酸菌a.orientalis中以提高其整合效率,简化遗传操作。
技术实现要素:
本发明的第一个目的是提供一种基于crispr/cas9编辑系统在东方拟无枝酸菌中进行基因敲除与敲入的方法,该方法将crispr/cas9编辑质粒plyny04引入东方拟无枝酸菌amycolatopsisorientalishccb10007中,同时实现了基因敲除与敲入,可以简化遗传操作,提高东方拟无枝酸菌的整合效率。本发明的第二个目的是提供一种含有gtfd基因同源臂的crispr/cas9编辑质粒plyny04,将egfp插入东方拟无枝酸菌的靶标序列,以便于检测基因编辑结果。本发明的第三个目的是提供一种含有gtfd基因同源臂的crispr/cas9编辑质粒plyny04在制备万古霉素高产菌的应用。本发明的第四个目的是提供一种含有gtfd基因同源臂的crispr/cas9编辑质粒plyny04在制备定点整合东方拟无枝酸菌突变株中的应用。本发明的第五个目的是提供一种能有效编辑东方拟无枝酸菌的万古霉素合成簇糖基转移酶gtfd基因的sgrna的引物对序列,所述引物对序列如seqidno:1和seqidno:2所示。
基于以上目的,本发明的第一个方面,提供一种基于crispr/cas9编辑系统在东方拟无枝酸菌中进行基因敲除与敲入的方法,包括如下步骤:
步骤一、构建含有cas9和sgrna的骨架质粒pkcpgcas9egdgtfd-na;
步骤二、构建含有egfp的靶标序列同源臂;
步骤三、含有gtfd基因同源臂的编辑质粒plyny04的构建;
将步骤一的骨架质粒pkcpgcas9egdgtfd-na使用限制性内切酶进行线性化;接着,使用同源重组试剂盒将步骤二的带有egfp的靶标序列同源臂整合到线性化的pkcpgcas9egdgtfd-na中,获得含有gtfd基因同源臂的编辑质粒plyny04;
步骤四、将编辑质粒plyny04电转化入a.orientalishccb10007
将步骤三得到的含有gtfd基因同源臂的编辑质粒plyny04电转入感受态a.orientalishccb10007细胞,同时实现egfp的敲入以及定点靶标序列的敲除。
根据本发明,所述含有cas9和sgrna的骨架质粒pkcpgcas9egdgtfd-na的构建方法为:利用ndei/hindiii双酶切pkccas9do,获得cas9sco载体;接着,以pkccas9do为模板,用如seqidno:1和seqidno:2所示的引物序列进行pcr扩增,获得sgrna重组片段;接着,以a.orientalishccb10007基因组为模板,使用如seqidno:3和seqidno:4所示的引物序列进行pcr扩增,获得内源gadph启动子;接着,以pib139为模板,用如seqidno:3和seqidno:4所示的引物序列进行pcr扩增,获得perme*启动子片段;接着,将cas9sco载体、sgrna重组片段、内源gadph启动子、perme*启动子片段连接,获得crispr/cas9骨架质粒pkcpgcas9egdgtfd-na。
根据本发明,含有egfp的靶标序列同源臂的构建方法为:以a.orientalishccb10007基因组作为模版分别用如seqidno:7和seqidno:8所示的引物序列以及用如seqidno:9和seqidno:10所示的引物序列扩增上下游同源臂片段;将扩增出的上下游产物经dna回收试剂盒回收后,采用如seqidno:11和seqidno:12所示的引物序列进行overlap重组,获得的gtfd同源臂片段经dna回收试剂盒回收后进行t/a克隆并测序,测序正确的质粒经kpni/psti酶切后回收;使用kpni/psti酶切质粒plygyq2,回收大小为1054bp的egfp序列,两个片段通过酶切连接获得带有egfp的gtfd同源臂。
本发明的第二个方面,提供一种含有gtfd基因同源臂的crispr/cas9编辑质粒plyny04,所述crispr/cas9编辑质粒plyny04是先采用上述方法构建获得骨架质粒pkcpgcas9egdgtfd-na和含有egfp的靶标序列同源臂,然后按上述方法将骨架质粒pkcpgcas9egdgtfd-na线性化,并使用同源重组试剂盒将带有egfp的gtfd同源臂整合到线性化的pkcpgcas9egdgtfd-na中构建获得。
本发明的第三个方面,提供一种含有gtfd基因同源臂的crispr/cas9编辑质粒plyny04在制备万古霉素高产菌的应用。
本发明的第四个方面,提供一种含有gtfd基因同源臂的crispr/cas9编辑质粒plyny04在制备定点整合东方拟无枝酸菌突变株中的应用。
本发明的第五个方面,提供一种能有效编辑东方拟无枝酸菌的万古霉素合成簇糖基转移酶gtfd基因的sgrna的引物对序列,所述引物对序列如seqidno:1和seqidno:2所示。
本发明的有益效果是:首次在东方拟无枝酸菌中构建crispr/cas9基因编辑系统,将egfp插入东方拟无枝酸菌的靶标序列,其作为报告基因更便于基因编辑结果的检测,通过荧光显微镜观测是否具有荧光即可。同时,本发明将crispr/cas9基因编辑系统引入东方拟无枝酸菌a.orientalis,相比以往的同源重组方法,整合效率得以大幅提高,同时简化了遗传操作。
附图说明
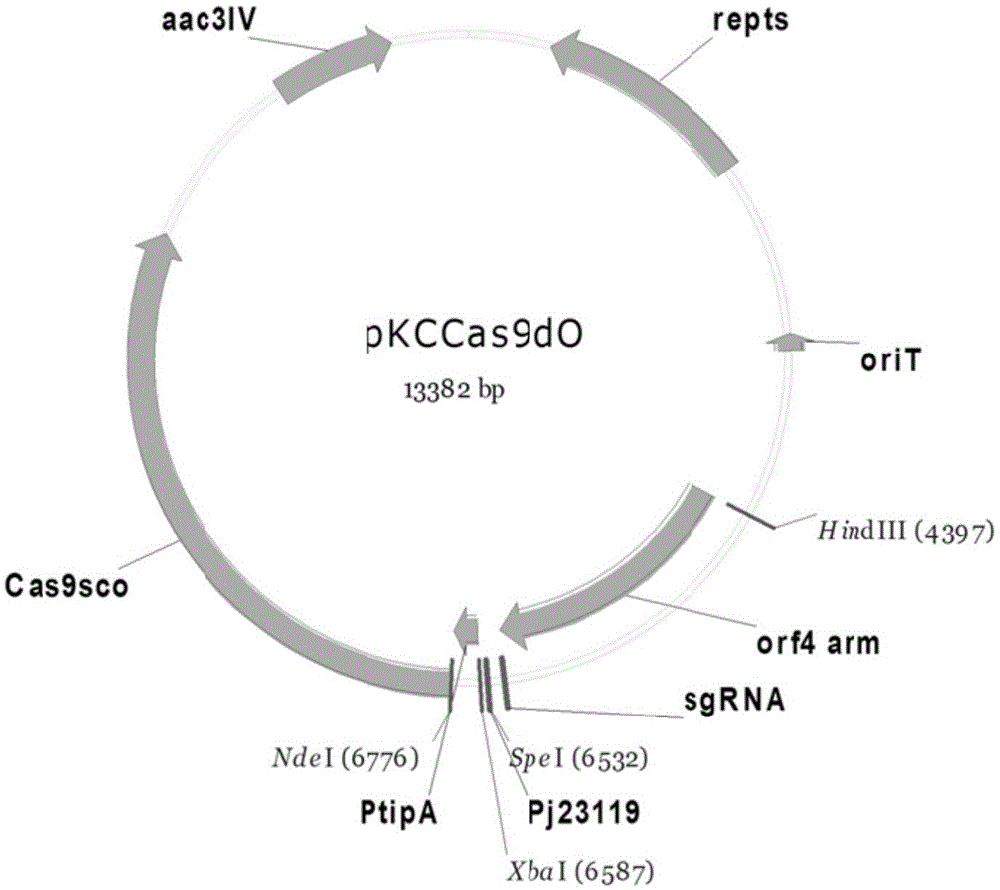
图1为pkccas9do质粒的结构示意图。
图2为pkcpgcas9egdgtfd-na骨架质粒的结构示意图。
图3为crispr/cas9编辑质粒plyny04的结构示意图。
图4为实施例5的基因敲除菌株的pcr验证图;其中,
泳道1为东方拟无枝酸菌染色体(阴性对照);泳道2为crispr/cas9编辑质粒plyny04(阳性对照);泳道3-7为阳性基因敲除菌株;泳道m为1kbdnaladdermarker(1kbdna分子量梯度标尺)。
a图为verify-gg-f/verify-gg-rpcr验证结果;b图为verify-g8-f/verify-g8-rpcr验证结果;c图为verify-10g-f/verify-10g-rpcr验证结果。
图5为实施例6的基因敲除菌株荧光检测图;其中,
a图为amycolatopsisorientalishccb10007荧光显微镜检测结果;b图为amycolatopsisorientalishccb10007非荧光显微镜检测结果;c图为基因敲除菌株荧光显微镜检测结果;d图为基因敲除菌株非荧光显微镜检测结果。
具体实施方式
以下结合具体实施例对本发明做进一步详细说明。应理解,以下实施例仅用于说明本发明而非用于限定本发明的范围。
以下实施例所用的试剂及质粒来源:
1、pkccas9do由中科院上海植物生理生态研究所构建,pkccas9do质粒的结构示意图如图1所示;
2、plygyq2由申请人自行构建(中国抗生素杂志,2014(3):193-197);
3、东方拟无枝酸菌(a.orientalis)hccb10007保藏于中国微生物菌种保藏管理委员会普通微生物中心,保藏号cgmccno.6023;
4、e.colijm110和pib139均为市售品。
实施例1质粒pkcpgcas9egdgtfd-na的构建和验证
步骤一、pkccas9do经ndei/hindiii双酶切获得cas9sco载体,酶切体系为:质粒2μl,ndei和hindiii内切酶各0.5μl,buffer1μl,补水至10μl。37℃水浴锅反应2h。
步骤二、sgrna重组片段的获得
以东方拟无枝酸菌amycolatopsisorientalishccb10007的万古霉素合成簇糖基转移酶gtfd基因为编辑对象设计sgrna,以pkccas9do为模板,用引物grnadnrecom和gtfdgrnarecompcr扩增获得sgrna重组片段,片段大小为103bp。
grnadnrecom:acgttgtaaaacgacggccagtgccaagcttctcaaaaaaagcaccgac(hindiii)
(seqidno:1)
gtfdgrnarecom:
ggcacaatcgtgccggttggtaggaactagtcgtcgagatcgcggtgtcgcgttttagagctagaaa(spei)(seqidno:2)
pcr反应体系为:10×reactionbuffer5μl,grnadnrecom和gtfdgrnarecom各1μl,pkccas9do1μl,dntps(2mm)0.5μl,koddnapolymerase(5u/μl)1μl,mg2+3μl,dmso2μl,ddh2o加至50μl。
pcr反应程序为:94℃热启动,5min;94℃变性,30s;退火温度68℃,30s;68℃延伸,kodplusneo酶系的效率为:1min/2kb,延伸10s。变性、退火、延伸三个步骤共30个循环。最后,68℃延伸10min,16℃放置。
步骤三、内源gadph启动子的获得
以a.orientalishccb10007基因组为模板,使用引物pgadphfw和pgadphrwpcr扩增,获得内源gadph启动子,片段大小为184bp;
pgadphfw:acactcgcatgcatactagagaatctctagaagctgagagttttcccaag(xbai)
(seqidno:3)
pgadphrw:ccaggccgatggagtacttcttgtccatatgtgctgccactcccttgaga(ndei)
(seqidno:4)步骤四、perme*启动子片段的获得
以pib139为模板,用引物ermefw和ermerwpcr扩增获得perme*启动子片段,片段大小为199bp。
ermefw:ggatttgttcagaacgctcggttgctctagagattctctagtatgcatgc(xbai)
(seqidno:5)
ermerw:cctaccaaccggcacgat(seqidno:6)步骤五、crispr/cas9骨架质粒pkcpgcas9egdgtfd-na的构建
将上述四个片段以gibson法进行连接,体系为:assembly缓冲液15μl,所有需要连接的片段5μl,将反应体系于pcr仪器中50℃保温60min。获得crispr/cas9骨架质粒pkcpgcas9egdgtfd-na,结构示意图见2。连接产物进行转化,导入感受态细胞dh5α,挑取单克隆于lb液体培养基中,37℃过夜培养,验证正确后提取质粒,备用。
实施例2crispr/cas9基因编辑系统同源臂的构建
步骤一、以a.orientalishccb10007基因组作为模版分别用vcm-8f/vcm-8r和vcm-10f/vcm-10r扩增上下游同源臂片段;
vcm-8f:aagcttagatcggtgagtcgctgctg(hindiii)(seqidno:7)
vcm8r:tcacgtatttccccgctgcagggtaccttcgctacccctgtttcgtg(psti)(kpni)
(seqidno:8)
vcm10f:aacaggggtagcgaaggtaccctgcagcggggaaatacgtgatgcgt(kpni)(psti)
(seqidno:9)
vcm-10r:aagcttttggtgatgatcaggcggga(hindiii)(seqidno:10)
步骤二、扩增出的上下游产物经dna回收试剂盒回收后进行overlap(重叠延伸pcr法)重组,获得的gtfd同源臂片段经dna回收试剂盒回收后进行t/a克隆并测序,测序正确的质粒经kpni/psti酶切后回收;
overlap引物序列为:
tycz-f:gtaaaacgacggccagtgccaagcttagatcggtgagtcgctgctg(hindiii)
(seqidno:11)
tycz-r:gagtcggtgctttttttgagaagcttttggtgatgatcaggcggga(hindiii)
(seqidno:12)
overlap重组pcr程序:引物使用上游片段的f引物、下游片段的r引物,延伸时间t为两个片段连接后大片段所需的时间。模板为上下游片段混合物。其余与普通pcr程序相同。
步骤三、使用与步骤二相同的限制性内切酶酶切质粒plygyq2,回收大小为1054bp的绿色荧光蛋白(egfp)序列,两个片段通过酶切连接获得带有绿色荧光蛋白的gtfd同源臂。
实施例3含有gtfd基因同源臂的crispr/cas9编辑质粒的构建
根据同源重组试剂盒说明书,将实施例2获得的带有绿色荧光蛋白的gtfd同源臂整合到pkcpgcas9egdgtfd-na中,获得含有gtfd基因同源臂的编辑质粒。进行具体操作如下:
步骤一、pkcpgcas9egdgtfd-na骨架质粒使用hindiii酶切线性化;
步骤二、根据公式计算重组反映所需要的dna量。(确保加样的准确性,各组分加样量不低于1μl)。
线性化载体:x=[0.02*克隆载体碱基对数]ng,插入片段:y=y1+y2…+yn=[0.04*每一个插入片段碱基对数]ng,2*clonexpressmix5μl,水加至10μl;
步骤三、配置反应体系;
步骤四、使用移液器轻轻吸打混匀(勿震荡混匀),短暂离心将反应液收集至管底,50℃,反应5min;降至4℃;
步骤五、连接反应结束后,将所有产物与50μl的dh5α感受态细胞吹吸混匀,以常规方法进行转化,涂布lb+相应抗性的lb固体平板,37℃倒置过夜培养。验证正确的即为crispr/cas9编辑质粒plyny04,结构示意图见图3。
实施例4将crispr/cas9编辑质粒电转化入a.orientalishccb10007
步骤一、将crispr/cas9编辑质粒plyny04转化入e.colijm110去甲基化;
步骤二、取2-5μg质粒于冰预冷的1.5mlep管中,加入60μlhccb10007感受态细胞,吹吸混匀后,立即放入预冷的电转杯(btx,φ2mm)中。电转化条件:600ω,25μf,7.5kv/cm,时长13毫秒。
步骤三、电转化后用1mltsb重悬菌体,转移至15ml玻璃试管中,于28℃振荡培养5~8小时。
步骤四、取100μl菌液涂布于含有相应抗生素的bennet平板培养基上,剩余的菌液离心浓缩后也涂相应的平板,28℃培养3~5天。
实施例5阳性菌株中crispr/cas9编辑质粒的消除及验证
挑选多株转化子,在含有apr抗性的3mltsb液体培养基中,28℃培养菌体2~3d。转化子菌液在无抗的本氏培养基划单菌落37℃过夜培养后,挑取单菌落对照涂布于无抗和含有apr的本氏培养基,28℃培养2-3天,无抗板上正常生长而有抗性板上不生长的单菌落即为成功消除质粒的阳性菌株。使用验证引物验证,结果如图4,使用的验证引物如下:
verify-g8-f:accggtcaaattgggcgtcg(seqidno:13)
verify-g8-r:gtgcagtgcttcagccgcta(seqidno:14)
verify-10g-f:gtgcagtgcttcagccgcta(seqidno:15)
verify-10g-r:atccaccggccgtccgccgc(seqidno:16)
verify-gg-f:gagttcgaggcgcgctatcg(seqidno:17)
verify-gg-r:accggagcatgtcctcggcc(seqidno:18)
实验结果:用引物verify-g8-f/verify-g8-r、verify-10g-f/verify-10g-r进行pcr,阳性转化子分别获得3340bp、3357bp的条带;用引物verify-gg-f/verify-gg-r进行,阳性对照和阳性转化子获得大小相同的条带,阴性对照获得与其大小不同的条带。
结论:已获得正确编辑的阳性菌株。
实施例6东方拟无枝酸菌绿色荧光蛋白的观察
对照菌株a.orientalishccb10007与阳性菌株均使用tsb液体培养基28℃培养4-5天。将菌株菌丝体涂于载玻片上面,先用普通光学显微镜观察菌丝是否生长正常并且拍照,再以共聚焦激光显微镜蓝光以40×的物镜观察绿色荧光蛋白的表达效果。结果如图5,获得的阳性菌株在共聚焦激光显微镜下可以明显检测到绿色荧光。
结果表明,编辑质粒plyny04成功地导入细胞中并完成了预期的同源重组,整合在其上的绿色荧光蛋白基因egfp获得有效表达。
实施例7gtfd基因敲除突变株发酵产物分析
发酵液中万古霉素及相关产物的含量分析参照2000版《美国药典/国家处方集》(usp24/nf19)中万古霉素的hplc分析方法进行。将出发菌株a.orientalishccb10007及gtfd基因敲除工程菌进行发酵后,hplc显示工程菌发酵液中已无万古霉素,进一步说明ccrispr/cas9成功敲除了万古霉素合成簇糖基转移酶gtfd基因。
以上所述,仅为本发明的较佳实施例,并非对本发明作任何形式上和实质上的限制,凡熟悉本专业的技术人员,在不脱离本发明技术方案范围内,当可利用以上所揭示的技术内容,而作出的些许更动、修饰与演变的等同变化,均为本发明的等效实施例;同时,凡依据本发明的实质技术对以上实施例所作的任何等同变化的更动、修饰与演变,均仍属于本发明的技术方案的范围内。
序列表
<110>上海交通大学
上海来益生物药物研究开发中心有限责任公司
<120>一种基因编辑方法在东方拟无枝酸菌中的应用
<130>181054
<141>2018-12-18
<160>18
<170>siposequencelisting1.0
<210>1
<211>49
<212>dna
<213>人工序列(artificialsequence)
<400>1
acgttgtaaaacgacggccagtgccaagcttctcaaaaaaagcaccgac49
<210>2
<211>67
<212>dna
<213>人工序列(artificialsequence)
<400>2
ggcacaatcgtgccggttggtaggaactagtcgtcgagatcgcggtgtcgcgttttagag60
ctagaaa67
<210>3
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>3
acactcgcatgcatactagagaatctctagaagctgagagttttcccaag50
<210>4
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>4
ccaggccgatggagtacttcttgtccatatgtgctgccactcccttgaga50
<210>5
<211>50
<212>dna
<213>人工序列(artificialsequence)
<400>5
ggatttgttcagaacgctcggttgctctagagattctctagtatgcatgc50
<210>6
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>6
cctaccaaccggcacgat18
<210>7
<211>26
<212>dna
<213>人工序列(artificialsequence)
<400>7
aagcttagatcggtgagtcgctgctg26
<210>8
<211>47
<212>dna
<213>人工序列(artificialsequence)
<400>8
tcacgtatttccccgctgcagggtaccttcgctacccctgtttcgtg47
<210>9
<211>47
<212>dna
<213>人工序列(artificialsequence)
<400>9
aacaggggtagcgaaggtaccctgcagcggggaaatacgtgatgcgt47
<210>10
<211>26
<212>dna
<213>人工序列(artificialsequence)
<400>10
aagcttttggtgatgatcaggcggga26
<210>11
<211>46
<212>dna
<213>人工序列(artificialsequence)
<400>11
gtaaaacgacggccagtgccaagcttagatcggtgagtcgctgctg46
<210>12
<211>46
<212>dna
<213>人工序列(artificialsequence)
<400>12
gagtcggtgctttttttgagaagcttttggtgatgatcaggcggga46
<210>13
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>13
accggtcaaattgggcgtcg20
<210>14
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>14
gtgcagtgcttcagccgcta20
<210>15
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>15
gtgcagtgcttcagccgcta20
<210>16
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>16
atccaccggccgtccgccgc20
<210>17
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>17
gagttcgaggcgcgctatcg20
<210>18
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>18
accggagcatgtcctcggcc20
- 还没有人留言评论。精彩留言会获得点赞!