一种基于双蒽的有机蓝色荧光材料及其制备方法和应用与流程
本发明属于有机电致发光二极管领域,具体涉及一种基于双蒽的有机蓝色荧光材料及其制备方法和应用。
背景技术:
有机电致发光二极管(organiclight-emittingdiodes,oled),因其全固态、自发光、广视角、低电压驱动、可柔性等特点,被认为是下一代显示和固态照明领域的最有竞争力的技术。在最近三十年,oled技术取得了突飞猛进的进步,其商业化进程也在不断地往前推进中。但总的来说,还存在有很多技术上的难题,但从材料的角度看,性能优越的红、绿、蓝光材料是实现全彩色显示的首要条件,但就目前的oled材料来看,除了绿光和红光的性能较好以外,蓝光材料仍不能满足商业化的生产,特别是深蓝光材料,由于其能隙大、效率低,还只能受限于部分领域的应用。最早应用于oled的蓝光材料就是蒽及其衍生物,但是由于其易结晶、成膜性差的缺点,大大降低了oled器件的效率及稳定性。因此,开发高效稳定的有机蓝色发光材料对推动oled的商业化进程具有更现实的意义。
技术实现要素:
本发明的目的在于克服上述现有技术的缺点,提供一种基于双蒽的有机蓝色荧光材料及其制备方法和应用;所制备的材料具有良好的热稳定性和较高的发光量子效率,应用于深蓝oled器件时,性能好、外量子效率(eqe)高。
为达到上述目的,本发明采用以下技术方案予以实现:
一种基于双蒽的有机蓝色荧光材料,其结构式如下:
其中,r1、r2为中性基团、供电子特性的基团或吸电子特性的基团。
本发明的进一步改进在于:
优选的,中性基团为h;供电子特性的基团为ch3、och3、c2h5、oc2h5或c(ch3)3;吸电子特性的基团为f、cf3、cn或cho。
一种上述基于双蒽的有机蓝色荧光材料的制备方法,采用9-苯蒽-10硼酸酯和1,3-二溴-4,5,6-三取代苯进行铃木反应制得,1,3-二溴-4,5,6-三取代苯中的取代基为r1和r2。
优选的,9-苯蒽-10硼酸酯通过9-溴-10苯蒽硼酸酯化制得,具体步骤为:将9-溴-10苯蒽、异丙氧硼酸酯、正丁基锂和thf按比例(1.4~2.8)g:(1.34~2.67)g:(0.42~0.81)g:(30~56)ml混合,在氮气或惰性气体氛围条件下,室温反应8~12小时,反应结束后通过萃取、旋蒸、柱层析和重结晶得到9-苯蒽-10硼酸酯。
优选的,正丁基锂的加入温度为-78℃。
优选的,9-溴-10苯蒽通过9-苯蒽溴化制得,具体制备步骤为:将9-苯蒽、n-溴代丁二酰亚胺和n,n-二甲基甲酰胺按(2.15~4.29)g:(1.8~3.6)g:(100~200)ml混合,在氮气或惰性气体氛围下,在85~90℃下反应1~2小时,甲醇洗涤并抽滤得到9-溴-10苯蒽。
优选的,9-苯蒽是9-溴蒽通过铃木反应得到的,具体制备步骤包括:将9-溴蒽、苯硼酸、四(三苯基膦)钯、甲苯、乙醇和k2co3溶液按(2.60~5.14)g:(1.83~3.66)g:(0.58~1.16)g:(100~200)ml:(30~60)ml:(30-60)ml混合,在氮气或惰性气体氛围条件下,100~110℃反应12~24小时后萃取,旋蒸,柱层析,重结晶得到9-苯蒽;所述k2co3溶液为13.82~27.64g的固体碳酸钾溶液加入至30~60ml水中配制而成。
优选的,铃木反应中,还加入有催化剂、溶剂和活化剂溶液,其中,9-苯蒽-10硼酸酯、1,3-二溴-4,5,6-三取代苯、催化剂、溶剂和活化剂之间的比例为(5~10)mmol:(1~2)mmol:(0.2~0.4)mmol:(50~100)ml:(22~44)mmol;铃木反应,是在氮气气体氛围条件下,在100~110℃温度下,反应12~24小时。
优选的,催化剂采用四(三苯基膦)钯;溶剂采用甲苯和乙醇的混合溶剂;活化剂采用k2co3。
一种上述基于双蒽的有机蓝色荧光材料在有机电致发光二极管器件中的应用。
与现有技术相比,本发明具有以下有益效果:
本发明公开了一种有机蓝色荧光材料,该材料是双蒽类衍生物,能够实现深蓝光发射。本发明采用苯作为中心π共轭桥链,在其间位上连接两个蒽发光单元,实现对有机材料分子的共轭形态的调控,实现材料分子的深蓝发光。同时,通过在中心苯π共轭桥链上引入具有供电子特性的基团或吸电子特性的基团,来有效抑制有机材料分子间的π-π堆积,实现材料高的发光量子效率。通过采用苯作为中心π共轭桥链,在其间位上连接两个蒽发光单元,调控有机分子的共轭形态、分子内电荷转移路径,以及实现材料分子的深蓝发射;同时,在中心π共轭桥链上引入空间位阻基团来消除有机材料分子间的π-π堆叠相互作用,实现高的发光量子效率;在非掺杂oled器件中,实现了深蓝光发射,色纯度高,性能优,能降低制作成本,简化生产工艺。总之,本发明的特点是:1)采用苯作为中心π共轭桥链,利用其两个间位来连接两个蒽发光单元,实现了对有机分子的共轭形态、激发态电荷转移及堆叠状态的调控;2)在中心π共轭桥链上引入具有供电子特性的基团或吸电子特性的基团,来进一步抑制有机分子间的π-π相互作用,提高材料分子的发光量子效率,实现深蓝发射。
本发明还公开了一种基于双蒽的有机蓝色荧光材料的制备方法,该方法以苯为π共轭桥链,在其间为上导入两个蒽发光单元,实现对有机材料分子的π共轭形态以及分子间π-π堆积方式的调控,实现在oled器件应用中深蓝光发射;同时,在中心π共轭桥链上引入具有供电子特性的基团或吸电子特性的基团,来进一步抑制有机材料分子间的π-π相互作用,从而提高材料分子的发光量子效率,成功的制备出好的热稳定性、高的荧光量子效率的有机深蓝荧光材料;材料合成步骤简单、产率高、易提纯。
本发明还公开了一种基于双蒽的有机蓝色荧光材料在有机电致发光二极管器件中的应用。制备出的有机蓝色荧光材料,在紫外光照射下,溶液或薄膜都呈现强的深蓝色荧光发射。以该有机蓝色荧光材料为主体发光材料,成功地制备出高性能非掺杂深蓝oled器件;该类材料应用在非掺杂oled中,cie的y值为0.08,实现了国家电视标准委员会(ntsc)的深蓝发射。
【附图说明】
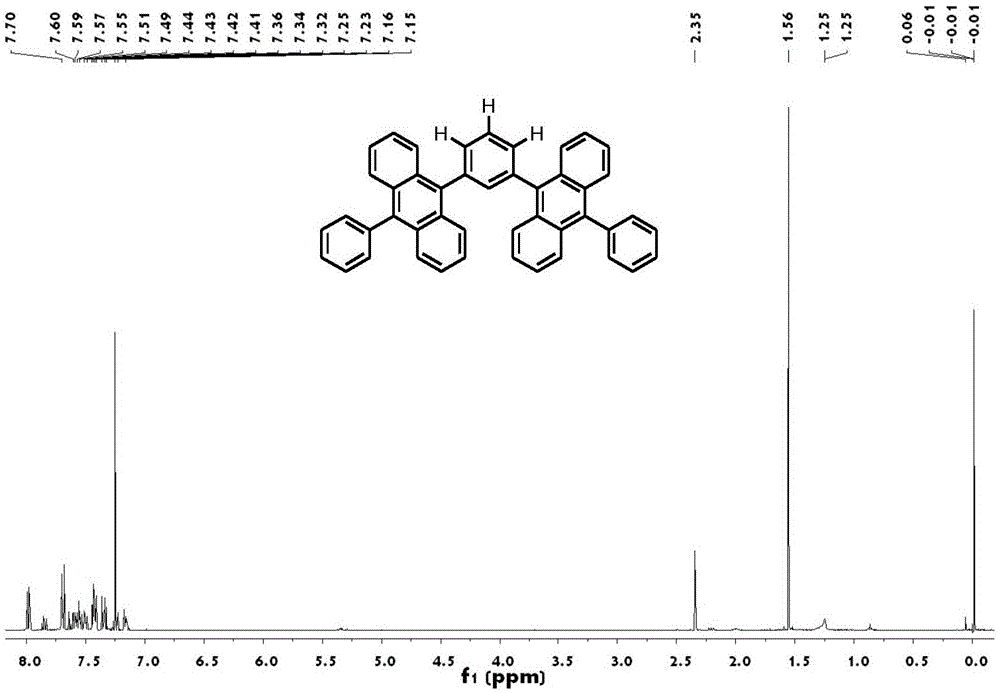
图1是本发明实施例1制得的有机蓝色荧光材料的核磁图谱。
图2是本发明实施例1制得的有机蓝色荧光材料的质谱图谱。
图3是本发明实施例1-3制得的蒽类衍生物m的热重图谱。
图4为本发明实施例1制得的有机蓝色荧光材料在不同溶液中的吸收光谱。
图5为本发明实施例1制得的有机蓝色荧光材料在不同溶液中的发射光谱。
图6为本发明实施例1制得的有机蓝色荧光材料在薄膜上的吸收和发射光谱。
图7为本发明制得的器件1的电致发光(el)图谱。
图8为本发明制得的器件1的电流密度-电压-亮度(cd-v-l)图谱曲线。
图9为本发明制得的器件1的电流效率-电流密度-功率效率(ce-cd-pe)的图谱曲线。
图10为本发明制得的器件1的电流密度-外部量子效率(cd-eqe)的图谱曲线。
【具体实施方式】
下面结合具体步骤和附图对本发明做进一步详细描述:
本发明公开了一种基于双蒽的有机蓝色荧光材料及其制备方法和应用;该发明通过对材料分子的精密设计,其中,以苯为π共轭桥链,在其间位上连接两个蒽分子发光单元,来调控有机分子的π共轭形态以及分子间的π-π堆叠作用,实现在oled器件应用中深蓝发光。同时,在中心π共轭桥链上导入供电子特性或吸电子特性的基团来进一步抑制有机分子之间的π-π堆叠作用,实现高的发光量子效率,制备出的有机蓝色荧光材料具有好的热稳定性和高的发光量子效率。其化学结构式如下。
其中r1、r2为中性基团或供电子特性的基团或吸电子特性的基团,中性基团为h;供电子特性的基团优选为ch3、och3、c2h5、oc2h5或c(ch3)3;吸电子特性的基团优选为f、cf3、cn或cho;合成出的有机蓝色荧光材料的化学结构式举例如下所示:
依次为化合物a、化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h、化合物i、化合物j、化合物m、化合物n、化合物o、化合物p;上述化合物为r1、r2中组合的任意一种,上述化学式只列举出一部分化合物,r1和r2的可选择基团详见表1。
表1r1和r2的可选基团
本发明的有机蓝色荧光材料的具体合成路线如下式(1)所示:
合成过程的具体过程包括以下步骤:
①制备9-苯蒽:将9-溴蒽2.60~5.14g,苯硼酸1.83~3.66g,碳酸钾溶液30~60ml,甲苯(toluene)100~200ml,乙醇(etoh)30~60ml加入到反应瓶,其中碳酸钾溶液是将13.82~27.64g碳酸钾加入30~60ml水中配比成的;最后加入0.58~1.16g四(三苯基膦)钯。然后对体系抽真空,在惰性气体或氮气保护下,100~110℃下回流12~24小时,进行suzuki偶联反应。反应结束后,萃取,旋蒸,柱层析,重结晶得到产物9-苯蒽。
②9-苯蒽的溴化:将9-苯蒽2.15~4.29g,n,n-二甲基甲酰胺(dmf)100~200ml,n-溴代丁二酰亚胺(nbs)1.8~3.6g加入到反应瓶,然后对体系抽真空,氮气或惰性气体保护下,85~90℃反应1~2小时。甲醇洗涤,抽滤得到产物9-溴-10苯蒽。
③9-溴-10苯蒽的硼酸酯化:将9-溴-10苯蒽1.40~2.80g,异丙氧硼酸酯1.34~2.67g,正丁基锂0.42~0.81g(在78℃下加入),thf30~56ml加入到反应瓶,然后对体系抽真空,氮气或惰性气体的保护下,室温下搅拌8~12小时。反应结束后,萃取,旋蒸,柱层析,重结晶得到产物9-苯蒽-10硼酸酯。
④终产物的合成:将9-苯蒽-10硼酸酯5~10mmol,1,3-二溴-4,5,6-三取代苯1~2mmol,催化剂四(三苯基)膦钯0.2~0.4mmol,甲苯40~80ml,乙醇10~20ml,k2co322~44mmol(用10~20ml蒸馏水配成的溶液),加入到反应瓶中;在氮气的保护下,在100~110℃回流12~24h进行suzuki偶联反应;反应结束后,萃取,旋蒸,柱层析,重结晶,升华得到产物;其中1,3-二溴-4,5,6-三取代苯中的取代基为供电子基团或吸电子基团;供电子特性的基团优选为h、ch3、och3、c2h5、oc2h5或c(ch3)3;吸电子特性的基团优选为f、cf3、cn或cho。
其中,1,3-二溴-4,5,6-三取代苯为1,3-二溴-5-甲基-4,6-二氢苯、1,3-二溴-5-氟-4,6-二氢苯、1,3-二溴-5-甲氧基-4,6-二氢苯、1,3-二溴-5-乙基-4,6-二氢苯、1,3-二溴-5-乙氧基-4,6-二氢苯、1,3-二溴-5-叔丁基-4,6-二氢苯、1,3-二溴-5-氰基-4,6-二氢苯、1,3-二溴-5-三氟甲基-4,6-二氢苯、1,3-二溴-5-醛基-4,6-二氢苯、1,3-二溴-5-氢-4,6-二甲氧基苯、1,3-二溴-5-氢-4,6-二甲基苯、1,3-二溴-5-氢-4,6-二醛基苯、1,3-二溴-5-氢-4,6-二氰基苯、1,3-二溴-5-氢-4,6-二乙基苯、1,3-二溴-5-氢-4,6-二乙氧基苯、1,3-二溴-5-氢-4,6-二叔丁基苯、1,3-二溴-5-氢-4,6-二氟苯、1,3-二溴-5-氢-4,6-二三氟苯、1,3-二溴-4,5,6-三甲氧基苯、1,3-二溴-4,5,6-三甲基苯、1,3-二溴-4,5,6-三氟苯、1,3-二溴-4,5,6-三氢苯、1,3-二溴-4,5,6-三乙氧基苯、1,3-二溴-4,5,6-三乙基苯、1,3-二溴-4,5,6-三叔丁基苯、1,3-二溴-4,5,6-三三氟苯、1,3-二溴-4,5,6-三氰基苯、1,3-二溴-4,5,6-三醛基苯。
上述化合物为r1和r2的部分组合,r1和r2能够自由匹配。
添加的碳酸钾溶液是为了活化硼酸根离子,使其转化为活性反应中间体。添加乙醇是为了增加溶剂的亲水性,使反应在均相条件下进行。
下面通过具体的实施例对本发明做进一步详细说明。
实施例1
①将9-溴蒽2.60g,苯硼酸1.83g,碳酸钾13.82g(加入30ml蒸馏水,配成2.0m溶液),甲苯100ml,乙醇30ml加入到反应瓶,再加入0.58g四(三苯基膦)钯。然后对体系抽真空,在氮气保护下,100℃回流12小时。反应结束后,甲苯萃取、旋蒸、柱层析(洗脱液:正己烷)、重结晶(正己烷/甲苯=4:1)得到产物。产率88%。
②9-苯蒽的溴化:将9-苯蒽2.15g,dmf100ml,nbs1.80g加入到反应瓶,然后对体系抽真空,在氮气保护下85℃反应1小时。反应结束后,甲醇洗涤,抽滤得到产物9-溴-10苯蒽。产率89%。
③9-溴-10苯蒽硼酸酯化:将9-溴-10苯蒽1.40g,异丙氧硼酸酯1.34g,正丁基锂0.42g(在-78℃下加入),thf30ml加入到反应瓶,对体系进行抽真空,在室温下搅拌8小时。反应结束后,萃取,柱层析(洗脱液正己烷/二氯甲烷=1:1),重结晶(正己烷/甲苯=3:1)得到产物9-苯蒽-10硼酸酯。产率60%。
④通过铃木(suzuki)偶联反应得到终产物m:将9-苯蒽-10硼酸酯5mmol,1,3-二溴-4,5,6-三氢苯1mmol,四(三苯基膦)钯0.2mmol,甲苯40ml,乙醇10ml,k2co322mmol(用10ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在100℃氮气的保护下回流12小时。反应结束后,柱层析(洗脱液:正己烷)、甲苯重结晶、升华得到终产物m。产率60%。核磁共振分析:nmr(400mhz,)δ=8.02–7.95(m,4h),7.88–7.81(m,1h),7.73–7.30(m,25h),7.28–7.21(m,5h),7.21–7.13(m,3h),2.35(s,3h),1.74–1.18(m,11h).核磁图谱如附图1所示。质谱(m/s),分子式为c46h30,理论值为582.23,实际值为582,质谱图谱如附图2所示。
实施例2
①将9-溴蒽3.50g,苯硼酸2.52g,碳酸钾18.82g(加入45ml水,配成溶液),甲苯136ml,乙醇45ml加入到反应瓶中,最后加入0.82g四(三苯基膦)钯。体系抽真空,氮气保护105℃条件下回流18小时。反应结束后,甲苯萃取、旋蒸、柱层析(洗脱液:正己烷)、重结晶(正己烷/甲苯=4:1)得到产物。产率86%。②9-苯蒽溴化:将9-苯蒽3.50g,dmf130ml,nbs2.94g加入到反应瓶,然后对体系抽真空,在氮气保护下88℃反应1.5h。反应结束后,甲醇热洗、抽滤,得到产物9-溴-10苯蒽,产率87%。③9-溴-10苯蒽硼酸酯化:将9-溴-10苯蒽2g,异丙氧硼酸酯1.91g,正丁基锂0.58g(在–78℃下加入),thf45ml加入到反应瓶,然后对体系抽真空,在室温下搅拌10小时。反应结束后,甲苯萃取、旋蒸、柱层析(洗脱液正己烷/二氯甲烷=1:1),重结晶(正己烷/甲苯=3:1)得到9-苯蒽-10硼酸酯产物,产率63%。④通过铃木(suzuki)偶联反应得到终产物m:将9-苯蒽-10硼酸酯8mmol,1,3-二溴-4,5,6-三氢苯1.6mmol,四(三苯基膦)钯0.32mmol,甲苯64ml,乙醇16ml,k2co335mmol(用16ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在105℃氮气的保护下回流20小时。反应结束后,柱层析(洗脱液:正己烷)、甲苯重结晶、升华得到终产物m。产率62%。质谱(m/s),分子式为c46h30,理论值为582.23,实际值为582。
实施例3
①将9-溴蒽5.14g,苯硼酸3.66g,碳酸钾27.64g(加入60ml水,配成溶液),甲苯200ml,乙醇60ml加入到反应瓶,最后加入1.16g四(三苯基膦)钯。然后对体系抽真空,在氩气保护下110℃回流24小时。反应结束后,甲苯萃取、旋蒸、柱层析(洗脱液:正己烷)、重结晶(正己烷/甲苯=4:1)得到产物。产率86%。②9-苯蒽溴化:将9-苯蒽4.29g,dmf200ml,nbs3.6g加入到反应瓶,然后对体系抽真空,在氩气保护下90℃反应2小时。反应结束后,甲醇热洗、抽滤,得到产物9-溴-10苯蒽,产率77%。③9-溴-10苯蒽硼酸酯化:将9-溴-10苯蒽2.80g,异丙氧硼酸酯2.67g,正丁基锂0.81g(在-78℃下加入),thf56ml加入到反应瓶,然后对体系抽真空,在室温下搅拌12小时。反应结束后,甲苯萃取、旋蒸、柱层析(洗脱液正己烷/二氯甲烷=1:1),重结晶(正己烷/甲苯=3:1)得到9-苯蒽-10硼酸酯产物,产率77%。④通过铃木(suzuki)偶联反应得到终产物m:将9-苯蒽-10硼酸酯10mmol,1,3-二溴-4,5,6-三氢苯2mmol,四(三苯基膦)钯0.4mmol,甲苯80ml,乙醇20ml,k2co344mmol(用20ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在110℃氩气的保护下回流24小时。反应结束后,柱层析(洗脱液:正己烷)、甲苯重结晶、升华得到终产物m。产率63%。质谱(m/s),分子式为c46h30,理论值为582.23,实际值为582。
实施例4
步骤①至步骤③同实施例1。
④通过铃木(suzuki)偶联反应得到终产物a:将9-苯蒽-10硼酸酯5mmol,1,3-二溴-4,6-二氢-5-甲基苯1mmol,四(三苯基膦)钯0.2mmol,甲苯40ml,乙醇10ml,k2co322mmol(用10ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在100℃氮气的保护下回流12小时。反应结束后,甲醇热洗抽滤、甲苯重结晶、升华得到终产物a。产率65%。质谱(m/s),分子式为c47h32,理论值为596.25,实际值为596。
实施例5
步骤①至步骤③同实施例3。
④通过铃木(suzuki)偶联反应得到终产物b:将9-苯蒽-10硼酸酯10mmol,1,3-二溴-4,6-二氢-5-氟苯2mmol,四(三苯基膦)钯0.4mmol,甲苯80ml,乙醇20ml,k2co344mmol(用20ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在110℃氮气的保护下回流24小时。反应结束后,甲醇热洗抽滤、甲苯重结晶、升华得到终产物b。产率68%。质谱(m/s),分子式为c46h29f,理论值为600.23,实际值为600。
实施例6
步骤①至步骤③同实施例3。
④通过铃木(suzuki)偶联反应得到终产物c:将9-苯蒽-10硼酸酯10mmol,1,3-二溴-4,6-二氢-5-甲氧基苯2mmol,四(三苯基膦)钯0.4mmol,甲苯80ml,乙醇20ml,k2co344mmol(用20ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在110℃氮气的保护下回流24小时。反应结束后,甲醇热洗抽滤、甲苯重结晶、升华得到终产物c。产率66%。质谱(m/s),分子式为c47h32o,理论值为612.25,实际值为612。
实施例7
步骤①至步骤③同实施例3。
④通过铃木(suzuki)偶联反应得到终产物d:将9-苯蒽-10硼酸酯10mmol,1,3-二溴-4,6-二氢-5-乙基苯2mmol,四(三苯基膦)钯0.4mmol,甲苯80ml,乙醇20ml,k2co344mmol(用20ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在110℃氮气的保护下回流24小时。反应结束后,甲醇热洗抽滤、甲苯重结晶、升华得到终产物d。产率66%。质谱(m/s),分子式为c48h34,理论值为610.27,实际值为610。
实施例8
步骤①至步骤③同实施例3。
④通过铃木(suzuki)偶联反应得到终产物e:将9-苯蒽-10硼酸酯10mmol,1,3-二溴-4,6-二氢-5-乙氧基苯2mmol,四(三苯基膦)钯0.4mmol,甲苯80ml,乙醇20ml,k2co344mmol(用20ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在110℃氮气的保护下回流24小时。反应结束后,甲醇热洗抽滤、甲苯重结晶、升华得到终产物e。产率66%。质谱(m/s),分子式为c48h34o,理论值为626.26,实际值为626。
实施例9
步骤①至步骤③同实施例3。
④通过铃木(suzuki)偶联反应得到终产物f:将9-苯蒽-10硼酸酯10mmol,1,3-二溴-4,6-二氢-5-叔丁基苯2mmol,四(三苯基膦)钯0.4mmol,甲苯80ml,乙醇20ml,k2co344mmol(用20ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在110℃氮气的保护下回流24小时。反应结束后,甲醇热洗抽滤、甲苯重结晶、升华得到终产物f。产率66%。质谱(m/s),分子式为c46h29,理论值为581.23,实际值为581。
实施例10
步骤①至步骤③同实施例3。
④通过铃木(suzuki)偶联反应得到终产物g:将9-苯蒽-10硼酸酯10mmol,1,3-二溴-4,6-二氢-5-氰基苯2mmol,四(三苯基膦)钯0.4mmol,甲苯80ml,乙醇20ml,k2co344mmol(用20ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在110℃氮气的保护下回流24小时。反应结束后,甲醇热洗抽滤、甲苯重结晶、升华得到终产物g。产率66%。质谱(m/s),分子式为c47h29n,理论值为607.23,实际值为607。
实施例11
步骤①至步骤③同实施例3。
④通过铃木(suzuki)偶联反应得到终产物h:将9-苯蒽-10硼酸酯10mmol,1,3-二溴-4,6-二氢-5-三氟甲基苯2mmol,四(三苯基膦)钯0.4mmol,甲苯80ml,乙醇20ml,k2co344mmol(用20ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在110℃氮气的保护下回流24小时。反应结束后,甲醇热洗抽滤、甲苯重结晶、升华得到终产物h。产率68%。质谱(m/s),分子式为c47h29f3,理论值为650.22,实际值为650。
实施例12
步骤①至步骤③同实施例3。
④通过铃木(suzuki)偶联反应得到终产物i:将9-苯蒽-10硼酸酯10mmol,1,3-二溴-4,6-二甲氧基-5-氢苯2mmol,四(三苯基膦)钯0.4mmol,甲苯80ml,乙醇20ml,k2co344mmol(用20ml蒸馏水配成溶液),加入到反应瓶,然后对体系抽真空,在110℃氮气的保护下回流24小时。反应结束后,甲醇热洗抽滤、甲苯重结晶、升华得到终产物i。产率68%。质谱(m/s),分子式为c48h34o2,理论值为642.26,实际值为642。
实施例13
步骤同实施例12。(只需将④中的1,3-二溴-4,6-二甲氧基-5-氢苯替换为1,3-二溴-4,6-二甲基-5-氢苯)合成的终产物为j。
实施例14
步骤同实施例12。(只需将④中的1,3-二溴-4,6-二甲氧基-5-氢苯替换为1,3-二溴-4,6-二醛基-5-氢苯)合成的终产物为k。
实施例15
步骤同实施例12。(只需将④中的1,3-二溴-4,6-二甲氧基-5-氢苯替换为1,3-二溴-4,6-二氰基-5-氢苯)合成的终产物为l。
实施例16
步骤同实施例12。(只需将④中的1,3-二溴-4,6-二氰基-5-氢苯替换为1,3-二溴-4,5,6-三甲氧基苯)合成的终产物为n。
实施例17
步骤同实施例16。(只需将④中的1,3-二溴-4,5,6-三甲氧基取代苯替换为1,3-二溴-4,5,6-三甲基取代苯)合成的终产物为o。
实施例18
步骤同实施例16。(只需将④中的1,3-二溴-4,5,6-三甲氧基苯替换为1,3-二溴-4,5,6-三氟苯)合成的终产物为p。
经测试,上述实施例1-3的目标产物m的热重图谱如附图3所示,从热重图谱可以得出m的热分解温度为415℃,说明材料m有很好的热稳定性。
经测试,上述实施例1-3的目标产物m分别在环己烷、甲苯及1,2-二氯乙烷溶液中(~10-6moll-1)和薄膜上的紫外吸收和荧光发射光谱:图4和图5分别实施例1-3制得的有机蓝色荧光材料为在不同溶液中的吸收和发射光谱;图6分别为实施例1-3制得的有机蓝色荧光材料在薄膜的吸收(正方形)和发射(圆圈)光谱:从图4可以得出,m在不同溶剂中的吸收峰分别为356/373/394、357/375/397、358/376/398nm,这些峰都是来自于蒽的特征吸收峰。从溶剂的极性由小到大未发生很大的变化,说明分子基态的偶极矩变化很小。从图5可以得出,m在不同溶剂中的发射峰分别为415、417、418nm,其中随着溶剂极性的增加有略微红移现象,归因于在激发态时分子内偶极矩微弱的变化。从图6可以得出,m的吸收和发射峰分别为359/379/403、427nm这些峰都是来自于蒽的特征吸收和发射峰。
应用例1
有机电致发光器件1的制备
本实施例按照下述方法制备有机电致发光器件1:
a)清洗ito(氧化铟锡)玻璃:分别用洗洁精,去离子水,thf,异丙醇溶剂超声清洗ito玻璃分别20分钟,然后在等离子体清洗器中处理10分钟,其方阻为15-20ω/sq;
b)在阳极ito玻璃上真空蒸镀空穴传输层npb,厚度为30nm;
c)在空穴传输层npb之上真空蒸镀电子阻挡层tcta,厚度为10nm。
d)在电子阻挡层tcta之上,真空蒸镀发光层化合物m,厚度为20nm;
e)在发光层之上,真空蒸镀作为电子传输层的tpbi,厚度为40nm;
f)在电子传输层之上,真空蒸镀电子注入层lif,厚度为1nm;
g)在电子注入层之上,真空蒸镀阴极al,厚度为100nm。
有机电致发光器件1的结构为依次叠层的ito(110nm)*/npb(30nm)/tcta(10nm)/eml:[m(20nm)]/tpbi(40nm)/lif(1nm)/al(100nm)。
其中器件1(发光层为化合物m)所使用的材料分子结构式如下表2所示:
表2器件1所使用的材料分子结构式
经测试,器件1的发光(el)图谱、电流密度-电压-亮度(cd-v-l)图谱曲线、电流密度-电流效率(cd-ce-pe)、电流密度-外部量子效率(cd-eqe)的图谱曲线分别如图7、图8、图9、图10所示:从图7可以得出:器件1是来自于材料m的发光,且在不同的电流密度下光谱不变,说明它的电致发光光谱有良好的稳定性。从图9可以得出:器件1的最大电流效率为1.19cd/a,从图9、10得出:器件1的效率滚降很小,说明器件效率稳定性好。
器件1的光电性能参数如下表3所示
表3电致发光器件1的光电参数
上表中,【a】器件名称,【b】发光层,【c】代表点亮电压,【d】代表最大电流效率,【e】代表最大功率效率,【f】代表最大外部量子效率,【g】代表在500cd/a时的最大外部量子效率,【h】代表色纯度坐标(cie)。
基于本发明材料的有机器件点亮电压为4.4v,实现了低电压驱动的非掺杂深蓝色oled器件。本发明的有机发光器件可以使用在电子照相感光体、平面显示器、复印机、打印机、液晶显示背光源、计时器等光源、以及各种发光器件、各种显示器件、各种标志、各种传感器、各种门牌等方面。
本发明所制备的材料合成成本低、产率高、易提纯等特点,在溶液或薄膜状态下都呈现强的深蓝色荧光发射。本发明所制备的材料应用在oled器件中,实现了蓝光发射。本发明所制备的材料在有机显示、有机固态照明等有机光电领域具有一定的应用价值和市场前景。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!