一种乌苯美司ε新晶型及其制备方法与用途与流程
本发明属于药物晶型
技术领域:
,特别涉及一种乌苯美司ε新晶型,并且涉及该乌苯美司ε新晶型的制备方法,还涉及该乌苯美司ε新晶型在制备口服固体制剂中的用途。
背景技术:
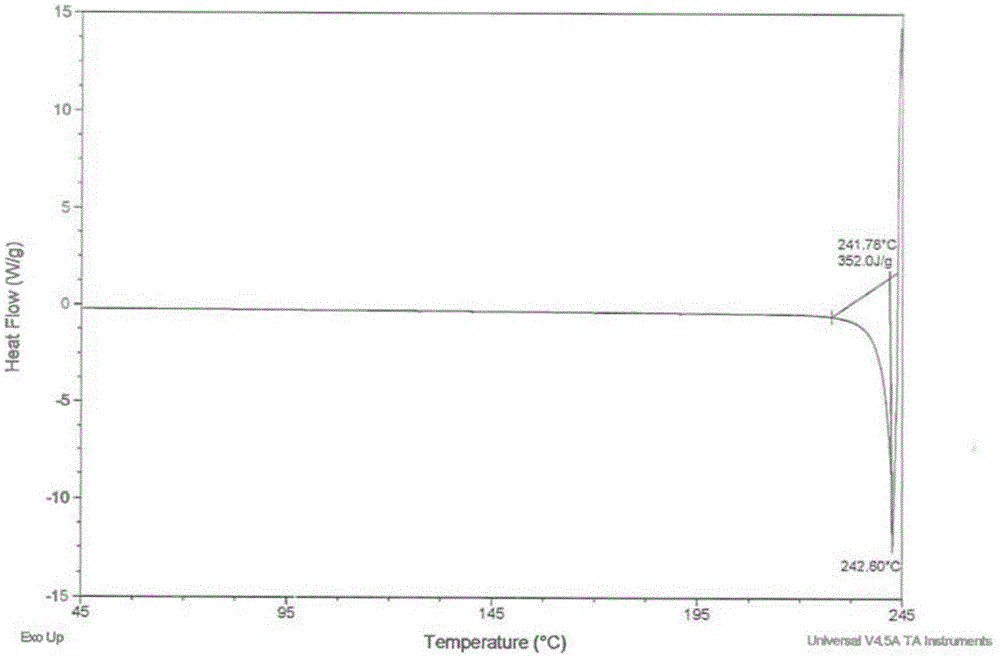
:乌苯美司(ubenimex),化学名为n-[(2s,3r)-3-氨基-2-羟基-1-氧代-4-苯基丁基]-l-亮氨酸,其化学结构式如下所示:乌苯美司最初是由日本科学家从链霉菌属(streptomycesofivoreticuli)的培养液中分离所得的二肽化合物,其可竞争性地抑制氨肽酶b(aminopeptidaseb)及亮氨酸肽酶(leucineaminopeptidase);增强t细胞的功能,使nk细胞的杀伤活力增强,且可使集落刺激因子合成增加而刺激骨髓细胞的再生及分化。因此,在临床上,乌苯美司能够增强免疫功能,用于抗癌化疗、放疗的辅助治疗,老年性免疫功能缺陷等;并且,其可配合化疗、放疗及联合应用于白血病、多发性骨髓瘤、骨髓增生异常综合症及造血干细胞移植后遗症,以及其他实体瘤患者。由于生物发酵的周期长、产率低,且占地面积大,故在目前原料药工业生产上,主要参照日本化药株式会社(nipponkayaku)hamaoumezawa等人的专利us4281180a和期刊论文《journalofantibiotics》1983,jun,36(6):695-9.所描述的工艺路线,通过化学合成方法获得乌苯美司。现有技术中,hamaoumezawa等人的专利ca1280550c描述了乌苯美司原料药的3种晶型:α、β、γ,其中γ晶型的熔点约为233℃(熔融并分解);且实施例详细列出了α→β、α→γ和β→γ的转晶实验操作。此外,论文《journalofantibiotics》1983,jun,36(6):695-9.所得乌苯美司的熔点为233℃~236℃(分解),其为γ晶型。中国专利cn101891647a在us4281180a和《journalofantibiotics》1983,jun,36(6):695-9.的基础上,对拆分和缩合这两步进行了优化,并在对最后一步催化氢化得到的乌苯美司醋酸盐,进行与hamaoumezawa等相同的精制操作后,最终得到的乌苯美司成品的熔点为233℃~236℃(分解),其为γ晶型。已失效的中国专利cn103333089a,参考了cn101891647a所公开的制备方法,先制得中间体n-[(2s,3r)-4-苯基-3-苄氧甲酰氨基-2-羟基丁酰]-l-亮氨酸苄酯粗品,然后重结晶纯化得到晶型纯品,再经与上述相同的催化氢化和精制操作,最终制得的乌苯美司成品的熔点为235℃~236℃,其为γ晶型。众所周知,乌苯美司γ晶型即原研制剂所用原料药的晶型。技术实现要素:本发明的发明人在乌苯美司制剂研发过程中,意外地获得了一种乌苯美司ε新晶型,其在正常储存条件下表现出了比现有晶型更好的稳定性,因此有利于乌苯美司原料药的长期储存;此外,通过一系列实验检测发现,由该ε新晶型制备的乌苯美司固体制剂表现出更好的总混粉末流动性与混合均匀性,以及更好的溶出度。具体地,本发明第一方面提供了一种乌苯美司ε新晶型,其x射线粉末衍射图中的衍射角2θ在3.14±0.2,6.42±0.2,12.94±0.2,15.91±0.2,16.25±0.2,18.54±0.2,19.54±0.2,20.22±0.2,23.68±0.2和26.18±0.2处存在特征峰值。优选地,采用差示扫描量热法测定的乌苯美司ε新晶型的熔点为:onsetpoint为241.78℃,peakpoint为242.60℃;其中,所述差示扫描量热法的测试条件包括:仪器:差示扫描量热仪dscq2000v24.11build124;升温条件:以10℃/min的速率从45℃升温至245℃。并且,本发明第二方面提供了一种第一方面所述的乌苯美司ε新晶型的制备方法,包括以下步骤:s1:将乌苯美司溶于盐酸溶液中;s2:使用活性炭脱色,过滤;s3:向滤液中加入碱液,调节ph值到5~6;s4:析晶过夜,抽滤,滤饼用丙酮打浆,再次抽滤;s5:取滤饼,真空干燥,即得所述乌苯美司ε新晶型。优选地,在上述制备方法的s1中,所述盐酸溶液的浓度为0.2至0.4mol/l;并且,所述乌苯美司与所述盐酸溶液的用量比为100g:1000ml。优选地,在上述制备方法的s2中,活性炭脱色的温度为65±5℃且持续时间为20~30分钟。优选地,在上述制备方法的s3中,所述碱液为稀氨水。进一步优选地,在上述制备方法的s3中,所述稀氨水的浓度为5%~15%。优选地,在上述制备方法的s4中,析晶的温度为-10℃至10℃。优选地,在上述制备方法的s5中,真空干燥的温度为80℃至100℃且持续时间为1~3小时。此外,本发明第三方面还提供了所述的乌苯美司ε新晶型在制备口服固体制剂中的用途。例如,所述乌苯美司ε新晶型可用于制备片剂。总之,本发明所提供的乌苯美司ε新晶型在正常储存条件下(30℃±2℃/65%rh±5%rh)具有很好的稳定性,不易降解,适合长期储存;并且,所述乌苯美司ε新晶型在片剂制剂过程中,改善了片剂总混粉末的流动性及混合均匀性,显著提高了成品的溶出度,从而有利于口服固体制剂产品的研发。附图说明图1为实施例7所得新晶型ε的晶体的差示扫描量热图(dsc);图2为实施例7所得新晶型ε的晶体的x射线粉末衍射图(xrpd);图3为实施例7所得新晶型ε的晶体的热重分析图(tga);图4为实施例1所得α晶型的差示扫描量热图(dsc);图5为实施例1所得α晶型的x射线粉末衍射图(xrpd);图6为实施例1所得α晶型的热重分析图(tga);图7为实施例2所得γ晶型的差示扫描量热图(dsc);图8为实施例2所得γ晶型的x射线粉末衍射图(xrpd);图9为实施例2所得γ晶型的热重分析图(tga);图10为实施例3所得γ晶型的差示扫描量热图(dsc);图11为实施例3所得γ晶型的x射线粉末衍射图(xrpd);图12为实施例3所得γ晶型的热重分析图(tga);图13为实施例4所得混晶(α+β)的差示扫描量热图(dsc);图14为实施例4所得混晶(α+β)的x射线粉末衍射图(xrpd);图15为实施例4所得混晶(α+β)的热重分析图(tga);图16为实施例5所得β晶型的差示扫描量热图(dsc);图17为实施例5所得β晶型的x射线粉末衍射图(xrpd);图18为实施例5所得β晶型的的热重分析图(tga);图19为实施例6所得新晶型ε的差示扫描量热图(dsc)。图20为实施例7所得新晶型ε的晶体放置1年后的差示扫描量热图(dsc);图21为实施例7所得新晶型ε的晶体放置1年后的x射线粉末衍射图(xrpd);图22为实施例7所得新晶型ε的晶体放置1年后的热重分析图(tga)。具体实施方式下面结合具体实施方式对本发明作进一步阐述,但本发明并不限于以下实施方式。仪器:差示扫描量热仪dscq2000v24.11build124;升温条件:以10℃/min的速率从45℃升温至245℃;在一个优选实施方式中,发明人采用以上测试条件测得乌苯美司ε新晶型的熔点为:onsetpoint为241.78℃,peakpoint为242.60℃(具体参见图1)。在一个优选实施方式中,发明人按照以下特定条件对乌苯美司ε新晶型实施了x射线粉末衍射:x射线管cu-ka靶;x射线波长:1.54056a;管压:40kv,管电流:40ma;扫描角度:3°~40°,扫描速度:0.1s/step。据此,所得到的乌苯美司ε新晶型的x射线粉末衍射检测结果如下表所示(参见图2):表1峰序号2θdbgheightheight%areaarea%fwhm13.13528.1582140274417.02500116.90.14826.41613.764318116148100.0147769100.00.149312.9356.83841206283.944183.00.107415.9105.56591421400.912570.90.137516.2515.44981331070.735932.40.513618.5454.78041591080.74580.30.065719.5374.5399158181111.2139899.50.125820.2244.38721551320.813970.90.162923.6823.75381531000.615731.10.2401026.1823.40081341320.810480.70.121在一个优选实施例中,乌苯美司ε新晶型的制备方法包括以下步骤:s1:将乌苯美司溶于盐酸溶液中;s2:使用活性炭脱色,过滤;s3:向滤液中加入碱液,调节ph值到5~6;s4:析晶过夜,抽滤,滤饼用丙酮打浆,再次抽滤;s5:取滤饼,真空干燥,即得所述乌苯美司ε新晶型。在一个优选实施例中,所述盐酸溶液的浓度为0.2至0.4mol/l;并且,所述乌苯美司与所述盐酸溶液的用量比为100g:1000ml。在一个优选实施例中,活性炭脱色的温度为65±5℃且持续时间为20~30分钟。在一个优选实施例中,所述碱液为稀氨水。在一个进一步优选的实施例中,所述稀氨水的浓度为5%~15%。值得补充说明的是,若氨水的浓度过大,则不容易控制滴定终点(等电点),并且会导致挥发更厉害,因此不利于节能环保和劳动保护;另一方面,若氨水浓度过稀(<5%),则需要加大氨水用量,因此需要换用更大的生产设备,从而导致生产能力利用率偏低,并且可能会有更多的乌苯美司溶于水相而被过滤除去,并最终造成成本上升。在一个进一步优选的实施例中,所述稀氨水的浓度为5%。在一个优选实施例中,析晶的温度为-10℃至10℃。在一个优选实施例中,真空干燥的温度为80℃至100℃且持续时间为1~3小时。值得补充说明的是,一般情况下,乌苯美司精制工艺中用到的溶剂为水和丙酮,其中,丙酮的沸点只有56℃,且为非质子型的非极性有机溶剂,在真空干燥条件下,比较容易除去;但是,水的沸点高达100℃,且为质子型的极性溶剂,在不引起乌苯美司热降解的前提下,干燥温度越高,则越易将残留水分降至质量标准规定的限度以下,从而降低乌苯美司原料药在后续储存过程中发生水解等降解反应的几率。在一个进一步优选的实施例中,真空干燥的温度为100℃。下列实施例中的各步骤如无特殊说明均为常规操作,各设备如无特殊说明均能从公开商业途径获得。下列实施例中所使用的差示扫描量热仪的型号为dscq2000v24.11build124,dsc方法为:以10℃/min的速率从45℃升温至245℃;下列实施例中所采用的x射线粉末衍射条件包括:x射线管cu-ka靶;x射线波长:管压:40kv,管电流:40ma,扫描角度:3°~40°,扫描速度:0.1s/step;下列实施例中所使用的tga检测仪器型号为netzschtg209f3240-20-382-l。实施例1根据专利ca1280550c的描述,参照专利us4281180a的example19和期刊论文《journalofantibiotics》1983,jun,36(6):695-9.的方法,制备乌苯美司α晶型。在2000ml的三颈瓶中加入100g乌苯美司,室温下加入1000ml的0.3mol/l盐酸溶液,水浴加热至50℃,搅拌全溶。加入5g药用活性炭,升温至65℃,搅拌脱色0.5小时,抽滤,滤液自然冷至室温后,冰水浴冷却,搅拌下滴加5%的稀氨水,控制反应瓶中的ph值在5~6,析出大量白色沉淀。抽滤,水洗再抽干至无水滴滴下,得到乌苯美司α晶型约85g。tga检测:①至190℃累计失重36.79%,这是由于样品表面的吸附水受热后蒸发离去,对应dtg曲线的峰值为66.6℃(这与dsc图谱中的吸热峰65.56℃基本吻合);②190~260℃失重5.18%,表明该样品含1个结晶水,为一水合物(乌苯美司分子量mw308,水分子量mw18,18/(308+18)×100%≈5.52%),对应dtg曲线的峰值为230.9℃(这与dsc图谱中的吸热峰225.06℃基本吻合);③最后受热分解(>260℃)。dsc检测:结合上述tga的检测结果,可知①吸热峰65.56℃为吸附水受热蒸发离去最快的温度);②该样品(α晶型晶体)的onsetpoint为100.60℃、peakpoint为106.81℃(吸热峰),表明α晶型晶体吸热熔融;③吸热峰225.06℃为样品中1个结晶水受热蒸发离去最快的温度。xrpd检测:乌苯美司α晶型的x射线粉末衍射图中衍射角2θ在4.95、9.96、14.09、14.96、18.96、20.02、22.41、24.70、25.10、27.08和29.45处具有特征峰。实施例2重复专利ca1280550c的example1,制备乌苯美司γ晶型。从上述实施例1所得乌苯美司α晶型中取出10g,在150℃加热3小时,得到样品白色固体5.7g。tga检测:①至240℃累计失重1.75%,这是由于经过加热干燥后,样品表面残留的吸附水受热后缓慢蒸发离去;②随后受热分解(>240℃),与专利ca1280550c描述的乌苯美司γ晶型在233℃熔融并分解基本吻合。dsc检测:该样品的onsetpoint为224.84℃、peakpoint为232.03℃(吸热峰),表明该晶型晶体吸热熔融,与专利ca1280550c描述的乌苯美司γ晶型的熔点(233℃)基本吻合,而且在该吸热熔融峰之前均未见其他的吸热或放热峰,这与专利ca1280550c对γ晶型热分析现象的描述一致,确认该样品为γ晶型。xrpd检测:乌苯美司γ晶型的x射线粉末衍射图中衍射角2θ在3.17、6.44、12.97、15.87、16.24、18.58、19.55、20.24、23.62和26.17处具有特征峰。注:因不同检测单位与人员在不同时间的制样手法、检测亦不同,故择优取向等因素会导致xrpd的吸收峰与专利ca1280550c描述存在微小差异。实施例3重复《journalofantibiotics》1983,jun,36(6):695-9.,制备乌苯美司γ晶型。从上述实施例1所得乌苯美司α晶型中取出10g,加入100ml丙酮,搅拌打浆2小时,抽滤,滤饼在室温下用p2o5真空干燥6小时,得到样品白色固体5.5g。tga检测:①至240℃累计失重1.28%,这是由于经过丙酮打浆并用p2o5真空干燥后,样品表面残留的吸附水受热后缓慢蒸发离去;②随后受热分解(>240℃),与《journalofantibiotics》1983,jun,36(6):695-9.描述的熔点233℃~236℃(分解)基本吻合,与专利ca1280550c描述的乌苯美司γ晶型在233℃熔融并分解基本吻合。注:这与实施例2所得γ晶型的检测数据基本一致。dsc检测:该样品的onsetpoint为232.17℃、peakpoint为234.78℃(吸热峰),表明该晶型晶体吸热熔融,与《journalofantibiotics》1983,jun,36(6):695-9.描述的熔点233℃~236℃(分解)吻合,与专利ca1280550c描述的乌苯美司γ晶型的熔点(233℃)吻合,而且在该吸热熔融峰之前均未见其他的吸热或放热峰,这与专利ca1280550c对γ晶型热分析现象的描述一致,确认该样品为γ晶型。注:这与实施例2所得γ晶型的检测数据基本一致。xrpd检测:该晶型晶体的x射线粉末衍射图中衍射角2θ在3.25、6.53、13.06、16.01、16.37、18.66、19.65、20.28、23.74和26.30处具有特征峰。注:这与实施例2所得γ晶型的检测数据基本一致。实施例4参照专利ca1280550c的example2,制备乌苯美司β晶型。从上述实施例1所得乌苯美司α晶型中取出10g,在60℃加热3小时,得到样品白色固体8.1g。tga检测:①至160℃累计失重0.10%,这是由于经过加热干燥后,样品表面残留的吸附水受热后缓慢蒸发离去;②随后受热分解(>160℃)。dsc检测:该样品只有两个吸热熔融峰,peakpoint分别为102.84℃和222.86℃,表明这是混晶,这与专利cn105968026a指出的“us4786754a中直接加热可能出现混晶”的判断是吻合的。从实施例1中α晶型的dsc检测结果可知,该样品第一个吸热峰(102.84℃)应该是α晶型的熔融峰,而222.86℃则为β晶型的熔融峰。xrpd检测:该混晶的x射线粉末衍射图中衍射角2θ在3.22、6.49、13.02、15.98、18.62、19.60、20.25、26.24和32.64处具有特征峰。实施例5重复专利ca1280550c的example2。从上述实施例4所得乌苯美司α和β的混晶中取出5g,在50ml丙酮中室温搅拌1小时,抽滤,滤饼30℃减压干燥3h,得到样品白色固体3.5g。tga检测:①至200℃累计失重1.47%,这是由于经过丙酮打浆并真空干燥后,样品表面残留的吸附水受热后缓慢蒸发离去;②随后受热分解(>200℃)。dsc检测:该样品只有一个吸热熔融峰,onsetpoint为212.27℃、peakpoint为225.08℃,这与实施例4所得混晶中β晶型的吸热熔融温度(onsetpoint为210.10℃、peakpoint为222.86℃)基本吻合,且在该吸热熔融峰之前均未见其他的吸热或放热峰,故该样品应为纯β晶型。xrpd检测:该晶型晶体的x射线粉末衍射图中衍射角2θ在3.22、6.47、13.02、19.60、26.23和32.63处具有特征峰。实施例6制备乌苯美司ε新晶型从上述实施例1所得乌苯美司α晶型中取出10g,加入100ml丙酮,搅拌打浆2小时,抽滤,滤饼在80℃下真空干燥2小时,得到样品白色固体5.4g。dsc检测:该样品只有一个吸热熔融峰,onsetpoint为243.26℃、peakpoint为245.88℃。实施例7制备乌苯美司ε新晶型从上述实施例1所得乌苯美司α晶型中取出10g,加入100ml丙酮,搅拌打浆2小时,抽滤,滤饼在100℃下真空干燥2小时,得到样品白色固体5.4g。tga检测:①至240℃累计失重0.05%,这是由于经过丙酮打浆并高温真空干燥后,样品表面残留的吸附水受热后缓慢蒸发离去;②随后受热分解(>240℃)。在温湿度(30℃±2℃/65%rh±5%rh)下放置1年后的tga检测图谱基本未变。dsc检测:该样品只有一个吸热熔融峰,onsetpoint为241.78℃、peakpoint为242.60℃,该onsetpoint温度(241.78℃)与实施例6所得ε晶型onsetpoint温度(243.26℃)基本吻合(差值≤2℃)。在温湿度(30℃±2℃/65%rh±5%rh)下放置1年后的dsc检测图谱基本未变。xrpd检测:该晶型晶体的x射线粉末衍射图中衍射角2θ在3.14、6.42、12.94、15.91、16.25、18.54、19.54、20.22、23.68和26.18处具有特征峰。在温湿度(30℃±2℃/65%rh±5%rh)下放置1年后的xrpd检测图谱基本未变。实施例8制备乌苯美司ε新晶型从上述实施例1所得乌苯美司α晶型中取出10g,加入100ml丙酮,搅拌打浆3小时,抽滤,滤饼在100℃下真空干燥2小时,得到样品白色固体5.4g。dsc检测:该样品只有一个吸热熔融峰,onsetpoint为241.79℃、peakpoint为242.50℃。实施例9制备乌苯美司ε新晶型从上述实施例1所得乌苯美司α晶型中取出10g,加入100ml丙酮,搅拌打浆2小时,抽滤,滤饼在100℃下真空干燥3小时,得到样品白色固体5.3g。dsc检测:该样品只有一个吸热熔融峰,onsetpoint为241.84℃、peakpoint为243.0℃。实施例10乌苯美司ε新晶型与γ晶型的稳定性对比试验在温度30℃±2℃和湿度65%rh±5%rh下放置1年的色谱纯度、最大单杂对比表如下所示:表2分析以上数据,可知:放置1年后,乌苯美司ε新晶型样品的色谱纯度、最大单杂几乎未变;而乌苯美司γ晶型样品虽然最大单杂未变,但色谱纯度有所下降,说明乌苯美司已降解出了一些微小杂质。由此可见,ε晶型比γ晶型更稳定。实施例11乌苯美司ε新晶型晶体与γ晶型的制剂对比试验采用2种晶型的原料药,制备成乌苯美司片,对制备过程及产品的质量进行比较,以评估2种晶型对制剂产品的影响。片剂的具体处方如下表所示:表3名称每片处方量(mg)批处方量(20000片,g)乌苯美司10200微晶纤维素601200磷酸氢钙34680低取代羟丙基纤维素10200硬脂酸镁120总计1152300并且,片剂的制备工艺步骤如下:①粘合剂配制:称取处方量的淀粉,将淀粉缓缓加入不断搅拌的冷水中,再加入热水,继续搅拌至混合均匀即可。②混合:分别称取处方量的乌苯美司(2种晶型)、微晶纤维素、磷酸氢钙、低取代羟丙基纤维素,用湿法制粒机进行混合。③制粒:加入粘合剂制软材,过24目筛制粒。④干燥:将湿颗粒在60℃条件下进行干燥,水分控制在1%~3%。⑤整粒:干颗粒过24目筛网整粒。⑥总混:将干颗粒加入实验型v型混合机中,加入硬脂酸镁,混合5分钟。⑦压片:将总混颗粒加入料斗中,适当调整填料深度及片厚,使片重控制在110~120mg,硬度控制在8kg~10kg。关键指标检测结果对比:表4由上表对比数据可见,所述乌苯美司ε新晶型晶体在片剂制剂过程中,改善了片剂总混粉末的流动性及混合均匀性,显著提高了成品的溶出度。以上对本发明的具体实施例进行了详细描述,但其只是作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1