一种具有抗肿瘤活性的鬼臼毒素衍生物及其制备方法与流程
本发明涉及药物合成
技术领域:
,具体为一种具有抗肿瘤活性的鬼臼毒素衍生物及其制备方法。
背景技术:
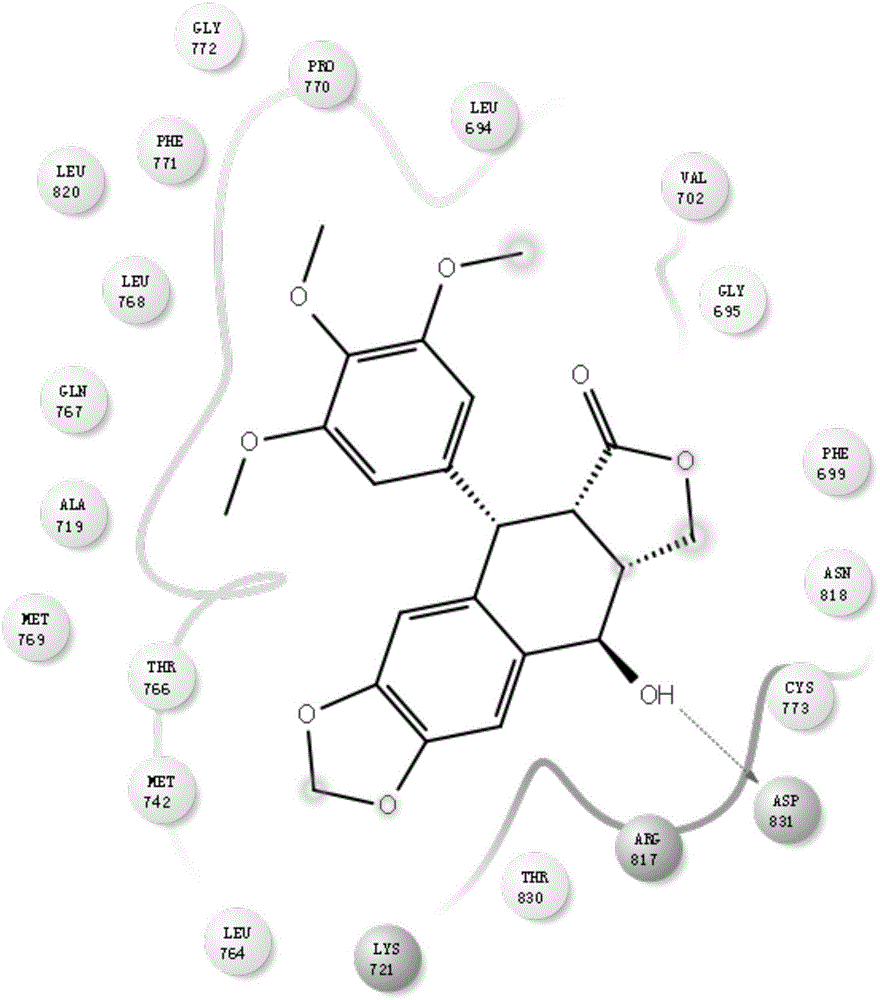
:鬼臼毒素存在于小檗科多年生草本类群鬼臼亚科、山荷叶属、八角莲属、足叶草属和桃儿七属植物的根和茎中,其具有的抗肿瘤作用得到广泛关注,研究者对鬼臼毒素进行结构改造并研制出依托泊苷和替尼泊苷等临床一线抗肿瘤药物,其衍生物如nk-611、gl331等也进入药物临床前研究,虽然现有的已合成的鬼臼毒素类衍生物具有抗肿瘤活性,但是存在水溶性差的问题,导致其形成的制剂在体内的利用度较低的缺陷,不利于制剂的生产及制备。技术实现要素:鉴于此,本发明提供了一种具有抗肿瘤活性的鬼臼毒素衍生物及其制备方法,提供了一种结构新颖的鬼臼毒素衍生物及该鬼臼毒素衍生物的制备方法,该鬼臼毒素衍生物对正常细胞具有安全性,对肿瘤细胞具有明显的抑制作用;该鬼臼毒素衍生物的制备方法简单、安全、易于操作和控制。本发明的技术方案如下:一种具有抗肿瘤活性的鬼臼毒素衍生物,其化学结构式为:制备上述具有抗肿瘤活性的鬼臼毒素衍生物的方法,步骤如下:(1)把鬼臼毒素溶于四氢呋喃中,将反应温度降至-20℃,缓慢滴加二异丙基氨基锂的四氢呋喃溶液,滴加完后降温至-40℃,缓慢滴加羰基提供化合物,滴加完后,降温至-60℃,在-60℃条件下反应20min,然后升温至-40℃,在-40℃条件下反应20min,然后升至0℃,在0℃条件下反应30min,最后倒入冰水中,缓慢滴加稀盐酸溶液,调节反应液ph为中性,然后用二氯甲烷萃取反应液多次,浓缩反应液后得到6-醛基-鬼臼毒素;其中,鬼臼毒素与二异丙基氨基锂和羰基提供化合物的投料量摩尔比为1:2:10;(2)方法一:将6-醛基-鬼臼毒素和叠氮化钠加入到氯仿中,再缓慢滴加三氟乙酸,滴加完后缓慢升至室温,在室温条件下反应至原料反应完全,缓慢加入饱和碳酸氢钠溶液,调节反应液ph为中性,分出有机相,有机相再经饱和氯化钠溶液洗涤,水相用氯仿萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到4-叠氮-6-醛基-鬼臼毒素;方法二:将6-醛基-鬼臼毒素和tms-n3加入到二氯甲烷中,滴加完后缓慢升至室温,在室温条件下反应原料反应完全,分出有机相,有机相再经饱和氯化钠溶液洗涤,水相用氯仿萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到4-叠氮-6-醛基-鬼臼毒素;4-叠氮-6-醛基-鬼臼毒素的制备方法为方法一、方法二的任一种;(3)在高压釜中,把4-叠氮-6-醛基-鬼臼毒素和脱水剂加入四氢呋喃中,再向反应釜内通入氨气,使反应釜内的压力得到0.05mpa,在50℃条件下反应一段时间,反应结束后,向反应液中滴加稀盐酸溶液,固体析出,抽滤反应液,将滤饼加入二氯甲烷中,然后用碳酸钾溶液调节二氯甲烷溶液的ph为中性,至固体完全溶解,再用无水硫酸钠干燥反应液,然后对反应液抽滤,将抽滤液浓缩得到4-叠氮-6-亚甲氨基-鬼臼毒素;其中,4-叠氮-6-醛基-鬼臼毒素与脱水剂的投料量质量比为1:2;(4)把4-叠氮-6-亚甲氨基-鬼臼毒素加入到丙酮中,再加入铁基催化剂,加热回流至原料反应完全,过滤反应液;在减压条件下,对过滤后的反应液进行浓缩,去除溶剂四氢呋喃,然后将浓缩后的反应液进行硅胶柱层析,分离获得纯净的8-(3,4,5-三甲氧苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮;(5)把8-(3,4,5-三甲氧苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮放入四氢呋喃中,搅拌均匀后加入无水溴化锂,升温到30℃,搅拌均匀后溶液逐渐变澄清;加热回流至原料反应完全,停止加热,冷却至室温,水洗后减压旋蒸除去大部分四氢呋喃,获得浓缩液,向浓缩液中然后加入二氯甲烷,在室温条件下边搅拌边加入稀盐酸调节浓缩液的ph为3±0.5,然后会浓缩液进行萃取,水相用二氯甲烷萃取后合并有机相,将合并的有机相进行浓缩,浓缩后加入甲醇,有大量固体析出,继续浓缩至少量体积,再加入一定量甲醇,缓慢搅拌下,加热至回流搅拌一段时间,停止加热;油浴自然降至室温后用冰水浴冷却,搅拌后过滤,滤饼以冷甲醇洗涤,抽干后得到8-(3,4,5-三羟基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮;(6)在高压釜中,把8-(3,4,5-三羟基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮和pamam-oh树枝状聚合物加入到甲醇中,缓慢通入环氧乙烷,然后通入氮气使反应体系压强维持在0.2mpa,反应温度控制在80±0.5℃,保温反应5h后,立即降温终止反应,向反应液中滴加盐酸调节ph为4-5,再加入活性炭加热回流0.5h后,趁热过滤,将滤液冷却至10℃,在搅拌条件下结晶,然后进行过滤、真空干燥得到粗品;将得到的粗品加入正己烷,加热回流至完全溶解,加入活性炭,继续回流一段时间后,趁热抽滤,将滤液降至室温静置析晶,抽滤、干燥得到8-(3,4,5-三羟乙氧基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮。在上述具有抗肿瘤活性的鬼臼毒素衍生物的制备方法的步骤(1)中,所述的羰基提供化合物为n,n-二甲基甲酰胺、n,n-二乙基甲酰胺或n,n-二丁基甲酰胺。在上述具有抗肿瘤活性的鬼臼毒素衍生物的制备方法的步骤(3)中,所述的脱水剂为无水硫酸钠、无水硫酸镁、无水氯化钙或无水氧化铝。在上述具有抗肿瘤活性的鬼臼毒素衍生物的制备方法的步骤(4)中,所述的铁基催化剂为氯化亚铁、四氧化三铁或二氧化铁。在上述具有抗肿瘤活性的鬼臼毒素衍生物的制备方法的步骤(6)中,所述的8-(3,4,5-三羟基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮与pamam-oh树枝状聚合物的投料量质量比为20:1~2。上述具有抗肿瘤活性的鬼臼毒素衍生物在抗肿瘤中的应用。对比现有技术,本发明的有益效果在于:(1)提供了一种新的鬼臼毒素衍生物及其制备方法;(2)该鬼臼毒素衍生物易溶于水;(3)该鬼臼毒素衍生物对正常细胞具有安全性,对肿瘤细胞具有明显的抑制作用;(4)该鬼臼毒素衍生物的制备方法简单、安全、易于操作和控制。附图说明图1为鬼臼毒素分子与肿瘤细胞靶点蛋白结合示意图;图2为本发明提供的鬼臼毒素衍生物分子与肿瘤细胞靶点蛋白结合示意图。具体实施方式为了使本
技术领域:
的人员更好地理解本发明中的技术方案,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。本发明的实施例1-3提供了化合物6-醛基-鬼臼毒素的制备过程;实施例4-5提供了化合物4-叠氮-6-醛基-鬼臼毒素的制备过程;实施例6-9提供了化合物4-叠氮-6-亚甲氨基-鬼臼毒素的制备过程;实施例10-12提供了化合物8-(3,4,5-三甲氧苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮的制备过程;实施例13提供了化合物8-(3,4,5-三羟基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮的制备过程;实施例14提供了化合物8-(3,4,5-三羟乙氧基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮的制备过程;具体过程详见实施例1-14,如下:实施例1化合物6-醛基-鬼臼毒素的制备过程(1)在反应瓶中,把鬼臼毒素4g溶于四氢呋喃50ml中,反应温度降温至-20℃,缓慢滴加二异丙基氨基锂2g的四氢呋喃溶液30ml,滴加完后降温至-40℃,缓慢滴加n,n-二甲基甲酰胺7.3g,滴加完后,保持-60℃反应20min,在升温至-40℃反应20min,最后升至0℃反应30min,最后倒入冰水100ml中,在0℃条件下,缓慢滴加稀盐酸溶液,调节反应液ph为中性,然后用二氯甲烷萃取反应液多次,浓缩反应液后经硅胶柱层析分离得到6-醛基-鬼臼毒素2.9g;1hnmr(400mhz,cdcl3):δ9.96(s,1h),7.37(d,j=4.0hz,1h),7.31(d,j=4.0hz,1h),6.82(s,1h),6.33(s,2h),5.48(d,j=8.0hz,1h),4.76(dd,j1=4.0hz,j2=4.0hz,1h),4.63(d,j=4.0hz,1h),4.37(t,j1=8.0hz,j2=8.0hz,2h),4.21(dd,j1=12.0hz,j2=4.0hz,1h),3.62(s,6h),3.45(s,3h);hrms(esi):443.1375[m+h]+;实施例2化合物6-醛基-鬼臼毒素的制备过程(2)本实施例与实施例1的区别为使用n,n-二乙基甲酰胺10g替代实施例1中的n,n-二甲基甲酰胺7.3g,获得的6-醛基-鬼臼毒素为3.3g。实施例3化合物6-醛基-鬼臼毒素的制备过程(3)本实施例与实施例1的区别为使用n,n-二丁基甲酰胺16g替代实施例1中的n,n-二甲基甲酰胺7.3g,获得的6-醛基-鬼臼毒素为3.6g。实施例4化合物4-叠氮-6-醛基-鬼臼毒素的制备过程(1)在0℃条件下,将6-醛基-鬼臼毒素4.5g和叠氮化钠1.3g加入到氯仿50ml中,再缓慢滴加三氟乙酸20ml,滴加完后缓慢升至室温,在室温条件下反应8h,tlc监控原料反应完全,缓慢加入饱和碳酸氢钠溶液,调节反应液ph为中性,分出有机相,有机相再经饱和氯化钠溶液洗涤,水相用氯仿萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到4-叠氮-6-醛基-鬼臼毒素3.5g;1hnmr(400mhz,cdcl3):δ9.93(s,1h),7.38-7.37(m,1h),7.31(d,j=4.0hz,1h),6.82(s,1h),6.31(s,2h),5.46(d,j=8.0hz,1h),4.76(dd,j1=4.0hz,j2=4.0hz,1h),4.63(d,j=4.0hz,1h),4.37(t,j1=8.0hz,j2=8.0hz,2h),4.25-4.24(m,1h),3.63(s,6h),3.47-3.46(m,3h);anal.calcdforc23h21n3o8:c,59.10;h,4.53;n,8.99.found:c,59.32;h,4.47;n,8.85。实施例5化合物4-叠氮-6-醛基-鬼臼毒素的制备过程(2)在室温条件下,将6-醛基-鬼臼毒素4.5g和tms-n31.2g加入到二氯甲烷50ml中,滴加完后缓慢升至室温,在室温条件下反应2h,tlc监控原料反应完全,分出有机相,有机相再经饱和氯化钠溶液洗涤,水相用氯仿萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到4-叠氮-6-醛基-鬼臼毒素4.1g;实施例6化合物4-叠氮-6-亚甲氨基-鬼臼毒素的制备过程(1)在高压釜中,把4-叠氮-6-醛基-鬼臼毒素4.6g和无水硫酸钠10g加入四氢呋喃100ml中,再向反应釜内通入氨气,使反应釜内的压力得到0.05mpa,在50℃条件下反应一段时间,反应结束后抽滤反应液,反应液中滴加稀盐酸溶液,逐渐由固体析出,抽滤反应液,滤饼加入二氯甲烷中,再用碳酸钾溶液调节反应ph为中性,固体完全溶解,再用无水硫酸钠干燥反应液,抽滤后浓缩反应液后经硅胶柱层析分离得到4-叠氮-6-亚甲氨基-鬼臼毒素2.3g;1hnmr(400mhz,cdcl3):δ8.87(s,1h),7.34(d,j=4.0hz,1h),7.29(d,j=4.0hz,1h),6.83(s,1h),6.27-6.25(s,2h),5.46(d,j=8.0hz,1h),4.74-4.73(m,1h),4.63(d,j=4.0hz,1h),4.37(t,j1=8.0hz,j2=8.0hz,2h),4.25-4.24(m,1h),3.63(s,6h),3.42(s,3h);anal.calcdforc23h22n4o7:c,59.22;h,4.75;n,12.01.found:c,59.04;h,4.83;n,12.16。实施例7化合物4-叠氮-6-亚甲氨基-鬼臼毒素的制备过程(2)本实施例与实施例6的区别为:使用无水硫酸镁10g替代实施例6中的无水硫酸钠10g,分离得到4-叠氮-6-亚甲氨基-鬼臼毒素为2.9g。实施例8化合物4-叠氮-6-亚甲氨基-鬼臼毒素的制备过程(3)本实施例与实施例6的区别为:使用无水氯化钙10g替代实施例6中的无水硫酸钠10g,分离得到4-叠氮-6-亚甲氨基-鬼臼毒素为3.4g。实施例9化合物4-叠氮-6-亚甲氨基-鬼臼毒素的制备过程(4)本实施例与实施例6的区别为:使用无水氧化铝10g替代实施例6中的无水硫酸钠10g,分离得到4-叠氮-6-亚甲氨基-鬼臼毒素为3.7g。实施例10化合物8-(3,4,5-三甲氧苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮的制备过程(1)把4-叠氮-6-亚甲氨基-鬼臼毒素4.6g加入到丙酮70ml中,在加入氯化亚铁1g,加热至回流,tlc监控原料反应完全,过滤反应液,滤液经减压条件下,蒸去绝大部分的溶剂四氢呋喃,经硅胶柱层析分离得纯净的8-(3,4,5-三甲氧苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮3.1g;1hnmr(400mhz,cdcl3):δ8.35(s,1h),7.11-7.09(m,1h),6.93(s,1h),6.17(s,2h),5.41(d,j=4.0hz,1h),4.68(d,j=4.0hz,1h),4.63(d,j=4.0hz,1h),4.37(t,j1=8.0hz,j2=8.0hz,2h),4.21(dd,j1=4.0hz,j2=12.0hz,1h),3.57(s,6h),3.39(s,3h);anal.calcdforc23h20n2o7:c,63.30;h,4.62;n,6.42.found:c,63.14;h,4.57;n,6.39。实施例11化合物8-(3,4,5-三甲氧苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮的制备过程(2)本实施例与实施例10的区别为:使用四氧化三铁1g替代实施例10中的氯化亚铁1g,分离得纯净的8-(3,4,5-三甲氧苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮为2.6g。实施例12化合物8-(3,4,5-三甲氧苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮的制备过程(3)本实施例与实施例10的区别为:使用二氧化铁1g替代实施例10中的氯化亚铁1g,分离得纯净的8-(3,4,5-三甲氧苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮为3.8g。实施例13化合物8-(3,4,5-三羟基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮的制备过程在500ml四口瓶中,把8-(3,4,5-三甲氧苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮43g在室温氩气保护下加入四氢呋喃600ml中,搅拌均匀后加入无水溴化锂34g,升温到到30℃,搅拌10min后溶液逐渐变澄清;缓慢升温至回流反应1.5h,大量固体产生,tlc检测反应原料消失;停止加热,冷却至室温,加入水600ml,搅拌20min后减压旋蒸除去大部分四氢呋喃,加入二氯甲烷600ml,室温搅拌下加入2n盐酸60ml至ph=3左右,萃取,分层,水相用二氯甲烷500ml萃取两次,合并有机相,水500ml洗涤两次,饱和食盐水300ml洗涤一次;有机相在40℃下浓缩旋出大部分二氯甲烷,加入甲醇100ml,有大量固体析出,继续浓缩至少量体积,加入甲醇400ml,缓慢搅拌下,加热至回流搅拌1h,停止加热;油浴自然降至室温后用冰水浴冷却,搅拌30min,过滤,滤饼以冷甲醇40ml洗涤多次,抽干后得到8-(3,4,5-三羟基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮33g,hrms(esi):395.0847[m+h]+。实施例14化合物8-(3,4,5-三羟乙氧基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮的制备过程在高压釜中,加入8-(3,4,5-三羟基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮40g、pamam-oh树枝状聚合物3g和甲醇300ml,缓慢通入环氧乙烷18g,然后通入氮气使反应体系压强维持在0.2mpa,反应温度控制在80℃,hplc监控反应,保温反应5h后,此时立即降温终止反应,滴加盐酸调节反应液ph为4-5,再加入2g活性炭加热回流0.5h后,趁热过滤,将滤液冷却至10℃,搅拌条件下结晶,经过滤后加入正己烷500ml,加热回流至完全溶解,加入针用活性炭3.0g,继续回流1.0h后,趁热抽滤,将滤液降至室温静置析晶2h后,抽滤、干燥得到本发明的鬼臼毒素衍生物44g,即8-(3,4,5-三羟乙氧基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮;1hnmr(400mhz,cdcl3):δ8.91(s,1h),7.22(d,j=12.0hz,1h),6.95(s,1h),6.22-6.20(m,2h),5.43(d,j=4.0hz,1h),4.66(d,j=4.0hz,1h),4.63(d,j=4.0hz,1h),4.57(d,j=8.0hz,4h),4.37(t,j1=8.0hz,j2=8.0hz,2h),4.31-4.29(m,2h),4.21(dd,j1=4.0hz,j2=12.0hz,1h),3.77(d,j=8.0hz,4h),3.18-3.15(m,2h);anal.calcdforc26h26n2o10:c,59.31;h,4.98;n,5.32.found:c,59.07;h,4.82;n,5.44。实施例15采用计算机药物辅助设计的方法,把鬼臼毒素和所得的鬼臼毒素衍生物分别与肿瘤细胞的靶点蛋白进行分子对接,靶点序号为1m17,从而发现,发现鬼臼毒素衍生物可以很好的进入靶点蛋白口袋里面,化合物分子被靶点蛋白残基氨基酸所包围,与靶点形成非常强的作用效果,鬼臼毒素分子中,只有羟基与天冬氨酸asp831形成氢键作用(如图1所示),而新化合物分子可以分别与天冬氨酸asp831,赖氨酸lys721,半胱氨酸cys773,天冬氨酸asp776和甲硫氨酸met769形成氢键作用(如图2所示)。实施例16抗肿瘤活性测试收集生长期肺癌细胞a549,以mts法测定本发明提供的鬼臼毒素衍生物的抗癌活性,将浓度为4×104个/ml的细胞溶液加到96孔细胞培养板中,其中,细胞培养板中含10%胎小牛血清得培养液配成单个细胞悬液,培养24h后,在37℃、体积浓度为5%的co2条件下与不同浓度的鬼臼毒素衍生物作用72h,然后将mts(最终质量浓度2mg/ml)和dms(最终摩尔浓度30μm)的混合物直接加入含细胞的培养基中,继续置培养箱孵育4h;作用4h后,弃去上清液,每孔加入150μldmso,振荡,细胞存活率通过其对mts作用的代谢物在酶联免疫监测仪490nm波长下的吸收率测定,鬼臼毒素衍生物对该细胞的抑制率ic50为26μmol·l-1。实施例17细胞毒性检测通过cck-8法检测不同浓度的目标化合物对人成纤维细胞存活率的影响,结果表明,不同浓度的目标化合物分别与细胞共同培养,各浓度组与阴性对照组相比较,差异无统计学意义,目标化合物对人成纤维细胞具有安全性。目标化合物浓度(μg/ml)02.55.07.510.0细胞存活率(%)100±094.391.987.281.6实施例18溶解度实验在250ml容量瓶中,加入林格氏液100ml,将鬼臼毒素和化合物8-(3,4,5-三羟乙氧基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮10mg、50mg、100mg、250mg、500mg、1000mg,分别加入容量瓶中,在超声波作用下观察是否溶解,发现鬼臼毒素10mg可以完全溶解,50mg部分溶解,化合物8-(3,4,5-三羟乙氧基苯基)-11,11a-二氢-8h-[1,3]二氧[4',5':5,6]苯并[1,2,3-de]呋喃[3,4-h]噌啉-9(8ah)-酮500mg可以完全溶解,1000mg部分溶解。以上实施例描述了本发明的基本原理、主要特征及优点,本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1