四环素分子印迹材料及其制备方法与流程
本发明涉及一种抗生素及其制备方法,特别是一种四环素表面分子印迹材料及其制备方法。
背景技术:
四环素是一类广谱类抗生素,常被应用于动物病菌类疾病的预防和治疗,在畜禽生产过程中所使用的四环素大部分以原药或代谢物的形式排泄到水体、土壤中,由于其母体化学结构稳定而不易分解,极易造成环境污染并诱导耐药菌产生。另外,可食用性动物组织如牛奶、肉类、蜂蜜中若残留高含量的四环素,长期食用可对人体造成消化机能失常、骨骼发育障碍及维生素缺乏的健康危害,因此世界各国都非常重视对四环素类tcs残留的检测。tcs目前常用的检测方法有微生物法、酶联免疫法、色谱法、毛细血管法。其中,微生物法测定所需时间长、专一性较低,酶联免疫法价格比较昂贵,而化学-物理方法则需要样品进行复杂的预处理过程。因此,需要研究一种简捷灵敏且具有高选择性的四环素类tcs残留检测方法。
分子印迹技术mit(molecularimprintingtechnology)是近年来发展起来的一种对某一目标化合物具有专一性识别的分子识别技术。采用分子印迹技术制备出的分子印迹聚合物具有立体专一识别性和通用性,其特殊形状和排列特定基团的纳米孔穴识别位点,能够通过特异性结合一种模板分子完成从复杂底物中分离富集某一特定痕量待测物的目的。因此,可通过制备四环素分子印迹材料用于痕量四环素的检测分析,现有技术的制备方法有本体聚合法、悬浮聚合法、沉淀聚合法。
niu等在硅胶表面制备了四环素分子印迹材料并与spe-hplc联用应用于牛奶样品中对四环素类兽药的富集分离和测定(文献[1]:牛玉玲等。用于选择性识别牛奶中四环素表面分子印迹材料的制备[j].中国食品工业,2014,(1):1-2.食品分析方法,2016,9(8):2342-2351.);sun等合成了四环素分子印迹整体柱,对其进行了色谱评价并应用于实际样品的分析(文献[2]:孙祥利等。分子印迹整体柱联用高效液相色谱法测定食品样品中四环素的研究,[j].食品工业,2009,79(3):926-934.)。王红涛研究了将分子印迹技术与电化学技术联合使用来检测水体中四环素(文献[3]:王红涛.基于分子印迹修饰电极的四环素选择性检测方法和仪器研究[d].大连理工大学,2011.)。现有技术的四环素分子印迹材料对模板分子容易产生“包埋”现象,印迹效率不高;且四环素分子印迹材料的识别位点位于聚合物内部,由于孔道深,印迹分子向聚合物内部扩散阻力较大,导致传质速度较慢、结合效率低的问题,这些问题均会对痕量四环素的检测分析效率和准确度产生影响。
技术实现要素:
本发明的目的是提供一种四环素分子印迹材料及其制备方法,要解决的技术问题是提高四环素的检测分析效率和准确度。
本发明采用以下技术方案:一种四环素分子印迹材料,为具有磁性的表面分子印迹聚合物m-mips,以fe3o4@tio2复合颗粒为载体,载体表面接枝有γ-(甲基丙烯酰氧)丙基三甲氧基硅烷mps,形成fe3o4@tio2-mps复合粒子,在fe3o4@tio2-mps复合粒子表面接枝有三氟甲基丙烯酸tfmaa,四环素分子印迹材料m-mips粒径为1~2μm,呈现微球状,其表面分布有与四环素空间结构相匹配、能识别四环素的孔穴;在纳米fe3o4颗粒表面包覆有介孔tio2,形成具有磁性的核壳结构的fe3o4@tio2复合颗粒。
本发明的fe3o4@tio2-mps复合粒子与三氟甲基丙烯酸tfmaa的质量比为1~2:5~7。
本发明的fe3o4@tio2复合颗粒与γ-(甲基丙烯酰氧)丙基三甲氧基硅烷mps的质量比为1~2:1~3。
本发明的四环素分子印迹材料对四环素的吸附量随四环素浓度的增加而增大,其最大吸附量为91.2mg/g。
在0~6min内四环素分子印迹材料对四环素的吸附量呈上升趋势,在6min之后,对四环素的吸附达到平衡。
本发明的四环素分子印迹材料对牛奶中四环素的加标回收率为98.3%。
一种四环素分子印迹材料的制备方法,包括以下步骤:
一、制备纳米fe3o4颗粒
室温下,将fecl3·6h2o溶于体积浓度为50%的乙醇中,fecl3·6h2o与乙醇质量比为:16~18:70~80,搅拌转速为200~400rmp,同时以2~5℃/min的升温速度,升温至30~60℃,按fecl3·6h2o与盐酸羟胺质量比:16~18:1~3,加入盐酸羟胺,搅拌反应5min后,用氨水调节ph至7~9,按fecl3·6h2o与油酸质量比为9:10,加入油酸,5~15min后,再以3~6℃/min的升温速度,升温至60~80℃,搅拌30min,自然冷却至室温,用磁铁将黑色沉淀分离出来,用20℃的无水乙醇洗涤,真空干燥,得到纳米fe3o4颗粒;
二、制备fe3o4@tio2复合颗粒
按钛酸丁酯tbt与无水乙醇体积比为1~2:6~8,将钛酸丁酯在无水乙醇中超声分散得到tbt分散液,按钛酸丁酯与盐酸摩尔比为1~2:0.01~0.02,将盐酸加入到tbt分散液中,在50~70℃下催化水解2~4h,空气中自然冷却至25℃,在转速200~400r/min,水浴温度25℃下,按钛酸丁酯与甘油体积比为1~2:5~10,加入甘油,封口,室温下静置凝胶15~20d,待凝胶干燥后真空干燥、研磨,然后用水作为溶剂索式抽提,真空干燥,得到介孔tio2;
按去离子水与无水乙醇体积比为1~3:12~15,无水乙醇与质量浓度为37%的浓盐酸体积比为12~18:0.1,配制成去离子水、无水乙醇、浓盐酸溶液;
按介孔tio2与无水乙醇体积比为1~3:3~4,将介孔tio2在无水乙醇中超声分散得到介孔tio2分散液,按纳米fe3o4颗粒与介孔tio2质量比为0.1~0.5:8~10,将纳米fe3o4颗粒放入三口瓶中,加入介孔tio2分散液,在40~60℃,转速200~400r/min下,水浴搅拌反应2~3min,溶胶,在纳米fe3o4颗粒表面包覆介孔tio2;
将去离子水、无水乙醇、浓盐酸溶液用恒压滴液漏斗,压力为88.35kpa,速度控制在0.1ml/s,逐滴加入至三口瓶中,室温下反应2h,凝胶,将反应产物转移至烧杯中静置6~12h,将沉淀物真空干燥,得到fe3o4@tio2复合颗粒;
三、制备fe3o4@tio2-mps复合粒子
按fe3o4@tio2复合颗粒与无水乙醇的质量与体积比0.4~0.6:50,将fe3o4@tio2复合颗粒超声分散于无水乙醇中,再按fe3o4@tio2复合颗粒与γ-甲基丙烯酰氧丙基三甲氧基硅烷(mps)质量比1~2:1~3,加入γ-甲基丙烯酰氧丙基三甲氧基硅烷(mps),加入0.5~1.0ml吡啶,在转速200~400r/min下,同时以3~6℃/min的升温速度,升温至30~50℃,搅拌反应24h,分离反应产物,用无水乙醇洗涤3次、砂心漏斗抽滤、真空干燥后,得到fe3o4@tio2-mps复合粒子;
四、制备fe3o4@tio2-mps-tfmaa复合粒子
按fe3o4@tio2-mps复合粒子与水质量与体积比1.0~3.0:120~180,将fe3o4@tio2-mps复合粒子加入水中,超声分散均匀后,在搅拌转速为200~400r/min条件下,以3~6℃/min的升温速度,升温至70℃,按质量比fe3o4@tio2-mps复合粒子:三氟甲基丙烯酸(tfmaa):过硫酸铵(nh4)2s2o8为1~2:5~7:0.02~0.05,先加入三氟甲基丙烯酸(tfmaa),充入氮气除氧20~30min,氮气流量为0.1l/min,再加入(nh4)2s2o8,接枝聚合反应24h后,在索式抽提器中用无水乙醇抽提24h,得到的固体物质,将固体物质在60℃、真空度为133pa条件下,干燥24h,得到fe3o4@tio2-mps-tfmaa复合粒子;
五、制备饱和吸附了四环素的复合粒子
将fe3o4@tio2-mps-tfmaa复合粒子1~3g,于40~50ml四环素溶液中,四环素溶液浓度为10mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比为6:7,室温下恒温振荡5~6h,振荡频率100r/min、功率为1680w,使fe3o4@tio2-mps-tfmaa复合粒子饱和吸附四环素,过滤出微粒,真空干燥,得到饱和吸附了四环素的复合粒子;
六、制备四环素分子印迹材料
将饱和吸附了四环素的复合粒子1~3g,于40~50ml四环素水溶液中,四环素水溶液浓度为4mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比为6:7,用naoh调节ph为7~8,按饱和吸附了四环素的复合粒子与交联剂质量比为1~2:1~2.5,加入交联剂乙二醇二缩水甘油(egde),搅拌转速为200~400r/min,同时以3~6℃/min的升温速度,升温至50℃,反应8h,反应结束后用水和甲醇在砂芯漏斗中抽滤、清洗至中性,在索氏提取器中用体积比为9:1的甲醇与冰乙酸混合溶液抽提24h,得到聚合物,然后用甲醇作为溶剂索式抽提,除去聚合物表面的乙酸,真空干燥,得到四环素分子印迹材料(m-mips)。
本发明方法的步骤三和步骤四中的质量与体积比为g与ml之比。
本发明的方法,进一步包括以下步骤:
一、制备纳米fe3o4粒子
室温下,将6.76gfecl3·6h2o溶于50%的乙醇中,fecl3·6h2o与乙醇质量比为17:75,转速为300rmp,同时以3℃/min的升温速度,升温至50℃,加入0.42g盐酸羟胺,搅拌反应5min后,用氨水调节ph至9,加入油酸8.3ml,10min后,再以4℃/min的升温速度,升温至70℃,搅拌30min,自然冷却至室温,用磁铁将黑色沉淀分离出来,用20℃的无水乙醇洗涤,真空干燥,得到纳米fe3o4颗粒;
二、制备fe3o4@tio2复合颗粒
按钛酸丁酯tbt与无水乙醇体积比为1:7,将钛酸丁酯在无水乙醇中超声分散得到tbt分散液,按钛酸丁酯与盐酸摩尔比为1:0.01,将盐酸加入到tbt分散液中,在60℃下催化水解3h,空气中自然冷却至25℃,在转速300r/min,水浴温度25℃下,按钛酸丁酯与甘油体积比为1:8,加入甘油,封口,室温下静置凝胶18d,待凝胶干燥后真空干燥、研磨,然后用水作为溶剂索式抽提,再真空干燥,得到介孔tio2;
将3ml去离子水、36ml无水乙醇、0.2ml浓盐酸配制成去离子水、无水乙醇、浓盐酸溶液。
将9g介孔tio2在36ml无水乙醇中超声分散得到介孔tio2分散液,将0.5g纳米fe3o4颗粒放入三口瓶中,加入介孔tio2分散液,在40℃,转速300r/min下,水浴搅拌反应2min,溶胶,在纳米fe3o4颗粒表面包覆介孔tio2;
将去离子水、无水乙醇、浓盐酸溶液用恒压滴液漏斗,压力为88.35kpa,速度控制在0.1ml/s,加入至三口瓶中,室温下反应2h,凝胶,将反应产物转移至烧杯中静置10h,将沉淀物真空干燥,得到fe3o4@tio2复合颗粒;
三、制备fe3o4@tio2-mps复合粒子
将0.5gfe3o4@tio2复合颗粒超声分散于50ml无水乙醇中,再加入0.625mlγ-甲基丙烯酰氧丙基三甲氧基硅烷(mps),加入1.0ml吡啶,在转速300r/min下,同时以4℃/min的升温速度,升温至50℃,搅拌反应24h后,用磁铁分离反应产物,用无水乙醇洗涤3次、砂心漏斗抽滤、真空干燥后,得到fe3o4@tio2-mps复合粒子;
四、制备fe3o4@tio2-mps-tfmaa复合粒子
将2.0gfe3o4@tio2-mps复合粒子,加入150ml水中超声分散均匀后,在搅拌转速为300r/min条件下,以4℃/min的升温速度,升温至70℃,先加入7ml三氟甲基丙烯酸(tfmaa),充入氮气除氧30min,氮气流量为0.1l/min,再加入0.042g(nh4)2s2o8,接枝聚合反应24h后,在索式抽提器中用无水乙醇抽提24h,得到的固体物质,将固体物质在60℃、真空度为133pa条件下,干燥24h,得到fe3o4@tio2-mps-tfmaa复合粒子;
五、制备饱和吸附了四环素的复合粒子
将2.0gfe3o4@tio2-mps-tfmaa复合粒子于50ml四环素溶液中,四环素溶液浓度为10mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比为6:7,振荡6h,振荡频率100r/min、功率为1680w,使fe3o4@tio2-mps-tfmaa复合粒子饱和吸附四环素,过滤出微粒,真空干燥,得到饱和吸附了四环素的复合粒子;
六、制备四环素分子印迹材料
将饱和吸附了四环素的复合粒子2.0g,于50ml四环素水溶液中,四环素水溶液浓度为4mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比为6:7,用naoh调节ph为8,加入2.5ml交联剂乙二醇二缩水甘油(egde),磁力搅拌,转速为300r/min,同时以4℃/min的升温速度,升温至50℃,反应8h,用水和甲醇在砂芯漏斗中抽滤、清洗至中性,在索氏提取器中用体积比为9:1的甲醇与冰乙酸混合溶液抽提24h,得到聚合物,然后用甲醇作为溶剂索式抽提,真空干燥,得到四环素分子印迹材料(m-mips)。
本发明与现有技术相比,具有磁性的表面分子印迹聚合物,吸附速度快,效率高,识别性能好,选择性高,吸附容量大,能够快速准确识别并分离食品样品中的四环素,具有较高选择性、回收率及可靠性。
附图说明
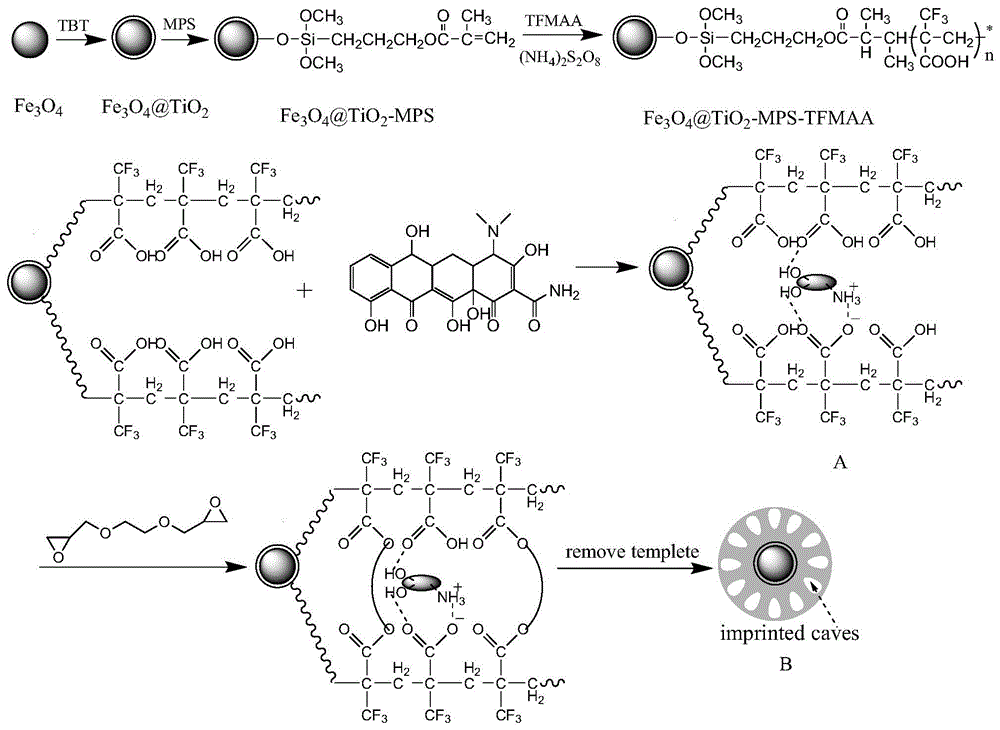
图1是本发明的制备方法反应过程示意图。
图2是本发明实施例2的纳米fe3o4颗粒透射电镜图片。
图3是本发明实施例2的m-mips扫描电镜图片。
图4是本发明实施例2的m-mips的吸附等温线。
图5是本发明实施例2的m-mips的吸附动力学曲线。
图6是本发明实施例2的m-mips分别与四环素、土霉素和金霉素的结合量示意图。
图7是本发明实施例2的m-mips固相萃取柱处理的色谱图。
具体实施方式
下面结合附图和实施例对本发明作进一步详细说明。
本发明的四环素分子印迹材料(分子印迹聚合物),为具有磁性的表面分子印迹聚合物m-mips,m-mips以fe3o4@tio2复合颗粒为载体,载体表面修饰接枝有γ-(甲基丙烯酰氧)丙基三甲氧基硅烷(mps),形成fe3o4@tio2-mps复合粒子,在fe3o4@tio2-mps复合粒子表面接枝有三氟甲基丙烯酸tfmaa,fe3o4@tio2-mps复合粒子与tfmaa的质量比为1~2:5~7,m-mips粒径为1~2μm,呈现微球状,m-mips表面分布有与四环素空间结构相匹配、能识别四环素的孔穴。
fe3o4@tio2复合颗粒与γ-甲基丙烯酰氧丙基三甲氧基硅烷mps的质量比为1~2:1~3。
fe3o4@tio2复合颗粒为在纳米fe3o4颗粒表面包覆有介孔tio2,形成具有磁性的核壳结构的fe3o4@tio2复合颗粒。
四环素分子印迹材料对四环素的吸附等温曲线,吸附量随四环素浓度的增加而增大,其最大吸附量为91.2mg/g,四环素分子印迹材料对四环素的吸附动力学曲线,0~6min内,四环素分子印迹材料对四环素的吸附量呈明显的上升趋势,在6min之后,对四环素的吸附达到平衡。
四环素分子印迹材料对牛奶中四环素的加标回收率为98.3%。
本发明的四环素分子印迹材料的制备方法,采用溶胶与凝胶方法在fe3o4表面包覆介孔tio2得到fe3o4@tio2复合颗粒,在复合颗粒表面接枝聚合双键得到fe3o4@tio2-mps,再以三氟甲基丙烯酸tfmaa为功能单体,乙二醇二缩水甘油醚egde为交联剂,制备具有磁性的表面分子印迹聚合物m-mips。如图1所示,包括以下步骤:
一、制备纳米fe3o4颗粒
室温(20℃)下,将fecl3·6h2o溶于体积浓度v/v为50%的乙醇中,fecl3·6h2o与乙醇质量比为:16~18:70~80,磁力搅拌,搅拌转速为200~400rmp,同时以2~5℃/min的升温速度,升温至30~60℃,持续搅拌下,按fecl3·6h2o与盐酸羟胺质量比:16~18:1~3,加入盐酸羟胺,搅拌反应5min后,用氨水调节ph至7~9,按fecl3·6h2o与油酸质量比为9:10,逐滴加入油酸,5~15min后,再以3~6℃/min的升温速度,升温至60~80℃,搅拌30min,反应结束,自然冷却至室温,用磁铁将黑色沉淀分离出来,并用20℃的无水乙醇洗涤,按现有技术真空干燥,得到纳米fe3o4颗粒。
纳米fe3o4颗粒使分子印迹聚合物具有磁性,在测定分子印迹聚合物的吸附性能时可以直接用磁铁将上清液与沉淀物分离,省去了离心这一步骤,简化了制备方法的工艺流程,测定的步骤。
二、制备fe3o4@tio2复合颗粒
按钛酸丁酯tbt与无水乙醇体积比为1~2:6~8,将钛酸丁酯在无水乙醇中按现有技术超声分散得到tbt分散液,按钛酸丁酯与盐酸摩尔比为1~2:0.01~0.02,将盐酸加入到tbt分散液中,直接在50~70℃下催化水解2~4h,空气中自然冷却至25℃,在转速200~400r/min,水浴温度25℃下,按钛酸丁酯与甘油体积比为1~2:5~10,加入模板溶液甘油,封口,室温下静置凝胶15~20d,待凝胶干燥后再按现有技术真空干燥、研磨,然后用水作为溶剂按现有技术索式抽提,除去模板溶液,再按现有技术真空干燥,得到介孔tio2。
按去离子水与无水乙醇体积比为1~3:12~15,无水乙醇与质量浓度为37%的浓盐酸体积比为12~18:0.1,配制成去离子水、无水乙醇、浓盐酸溶液待用。
按介孔tio2与无水乙醇体积比为1~3:3~4,将介孔tio2在无水乙醇中按现有技术超声分散得到介孔tio2分散液,按纳米fe3o4颗粒与介孔tio2质量比为0.1~0.5:8~10,将纳米fe3o4颗粒放入三口瓶中,加入介孔tio2分散液,在40~60℃,转速200~400r/min下,水浴搅拌反应2~3min,溶胶,在纳米fe3o4颗粒表面包覆介孔tio2。
将去离子水、无水乙醇、浓盐酸溶液用恒压滴液漏斗,压力为88.35kpa,速度控制在0.1ml/s,逐滴加入至三口瓶中,室温下反应2h,凝胶,将反应产物转移至烧杯中静置6~12h,将沉淀物按现有技术真空干燥,得到fe3o4@tio2复合颗粒。
fe3o4@tio2复合颗粒表达式中,“@”表示介孔tio2包覆在纳米fe3o4颗粒表面,形成核壳结构。本步骤采用溶胶-凝胶方法在fe3o4表面包覆介孔tio2,得到具有磁性的fe3o4@tio2核壳结构的复合颗粒,步骤简单、包覆率高。介孔tio2具有四方晶系的晶体结构,物理化学性质稳定,无毒无害,孔径小,比表面积大。fe3o4@tio2复合颗粒与纳米fe3o4颗粒相比,核壳结构导致m-mips有更好的稳定性,具有更大的吸附表面积,提高吸附四环素的性能,使吸附模板分子和解吸模板分子的速度更快,增大了吸附量和吸附速率。本步骤采用甘油表面修饰二氧化钛形成介孔tio2,进一步增大tio2比表面积,在fe3o4@tio2表面接枝mps时,增大接枝率。
三、制备fe3o4@tio2-mps复合粒子
按fe3o4@tio2复合颗粒与无水乙醇的质量g与体积ml比0.4~0.6:50,将fe3o4@tio2复合颗粒按现有技术超声分散于无水乙醇中,再按fe3o4@tio2复合颗粒与γ-甲基丙烯酰氧丙基三甲氧基硅烷mps质量比1~2:1~3,加入mps,加入0.5~1.0ml(约2~3滴)吡啶,在转速200~400r/min下,同时以3~6℃/min的升温速度,升温至30~50℃,机械搅拌反应24h后,用磁铁分离反应产物(沉淀物),将上清液与沉淀物分离,按现有技术用无水乙醇洗涤3次、砂心漏斗抽滤、真空干燥后,得到fe3o4@tio2-mps复合粒子。
fe3o4@tio2-mps复合粒子表达式中,“-”表示mps接枝聚合在fe3o4@tio2复合颗粒的表面。硅烷偶联剂mps与fe3o4@tio2表面以化学键方式结合,使mps接枝到fe3o4@tio2表面,在表面形成有机包覆层。mps在fe3o4@tio2-mps表面含有可聚合的双键,相当于充当了功能单体tfmaa形成的聚合物层与fe3o4@tio2-mps复合粒子之间的“桥梁”,便于将功能单体tfmaa接枝到fe3o4@tio2-mps复合粒子的表面,得到分子印迹聚合物的纳米级的聚合物层(分子印迹薄层)。
四、制备fe3o4@tio2-mps-tfmaa复合粒子
按fe3o4@tio2-mps复合粒子与水质量g与体积ml比1.0~3.0:120~180,将fe3o4@tio2-mps复合粒子加入水中,按现有技术超声分散均匀后,置于可加热磁力搅拌器上,在搅拌转速为200~400r/min条件下,以3~6℃/min的升温速度,升温至70℃,持续搅拌下,按质量比fe3o4@tio2-mps复合粒子:功能单体三氟甲基丙烯酸tfmaa:过硫酸铵(nh4)2s2o8为1~2:5~7:0.02~0.05,先加入tfmaa,充入氮气除氧20~30min,氮气流量为0.1l/min,再加入(nh4)2s2o8作为引发剂进行接枝聚合反应,接枝聚合反应24h后,在索式抽提器中用无水乙醇按现有技术抽提24h,得到的固体物质,将固体物质在60℃、真空度为133pa条件下,干燥24h,得到fe3o4@tio2-mps-tfmaa复合粒子。
fe3o4@tio2-mps-tfmaa复合粒子表达式中,第二个“-”表示tfmaa功能单体接枝在fe3o4@tio2-mps复合粒子的表面,fe3o4@tio2-mps表面的双键与tfmaa表面的双键发生聚合反应,使tfmaa连接到fe3o4@tio2-mps复合粒子上。在fe3o4@tio2-mps复合粒子表面接枝的tfmaa是为了其与模板分子四环素产生氢键、偶极-偶极或疏水相互作用的分子间作用力,使fe3o4@tio2-mps-tfmaa复合粒子对模板分子饱和吸附,形成模板分子的印迹。
五、制备饱和吸附了四环素的复合粒子
将fe3o4@tio2-mps-tfmaa复合粒子1~3g,于40~50ml四环素tc溶液中,tc溶液浓度为10mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比v/v为6:7,室温下恒温振荡5~6h,振荡频率100r/min、功率为1680w,使fe3o4@tio2-mps-tfmaa复合粒子饱和吸附tc,按现有技术过滤出微粒,真空干燥,得到饱和吸附了四环素的复合粒子。
如图1中的a部分所示,加入模板分子tc,饱和吸附了tc的tfmaa表面的官能团与模板分子tc的官能团产生氢键、偶极-偶极或疏水相互作用的分子间作用力,使tc被吸附在fe3o4@tio2-mps-tfmaa复合粒子表面。饱和吸附了四环素的复合粒子使模板分子四环素不溶于甲醇-乙腈溶剂,方便采用甲醇与冰乙酸混合溶液被洗脱下来,形成能识别四环素的孔穴。使最终制备得到的m-mips能更精确地检测到样品中的四环素。
六、制备四环素分子印迹材料
将饱和吸附了四环素的复合粒子1~3g,于40~50ml四环素水溶液中,四环素水溶液浓度为4mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比v/v为6:7,用naoh调节ph为7~8,按饱和吸附了四环素的复合粒子与交联剂egde质量比为1~2:1~2.5,加入交联剂乙二醇二缩水甘油egde,磁力搅拌,转速为200~400r/min,同时以3~6℃/min的升温速度,升温至50℃,反应8h,反应结束后按现有技术用水和甲醇在砂芯漏斗中抽滤、清洗至中性,在索氏提取器中用体积比v/v为9:1的甲醇与冰乙酸混合溶液按现有技术抽提24h,以除去模板分子四环素和未反应的交联剂egde,得到聚合物,然后用甲醇作为溶剂按现有技术索式抽提,除去聚合物表面的乙酸,再按现有技术真空干燥,得到四环素分子印迹材料m-mips。
加入egde,使饱和吸附在fe3o4@tio2-mps-tfmaa复合粒子表面的tc偶联在tfmaa表面,与其结合在一起,形成与四环素空间结构匹配的孔穴,用甲醇与冰乙酸混合溶液洗去模板分子tc后,在fe3o4@tio2-mps-tfmaa复合粒子表面保留形成的与tc空间结构匹配,官能团互补的孔穴,故m-mips表面形成有能够识别tc的特异性结合位点,使得制备得到的m-mips能更精确地检测到样品中的痕量tc,提高检测四环素的准确度。如图1中的b部分所示,四环素分子印迹材料m-mips为表面带有能识别四环素的孔穴、颗粒为纳米级、具有磁性的分子印迹聚合物。
本发明的方法制备得到的四环素分子印迹材料,用日立ht7800透射电子显微镜观测fe3o4复合粒子的亚显微结构或超微结构,用日本电子株式会社jeol的jsm-7200f型扫描电子显微镜观测分子印迹聚合物的表面形貌,用上海琅玕实验设备有限公司的shz-c水浴恒温振荡器测定分子印迹聚合物的吸附等温线和吸附选择性,用日本岛津公司的lc-20at型高效液相色谱仪和北京普析通用仪器有限责任公司的tu-1810紫外可见分光光度计测定其回收率。
四环素分子印迹材料的吸附等温线为以四环素浓度为横坐标,吸附量为纵坐标的曲线,用于测定分子印迹聚合物在不同四环素浓度时的吸附量。
四环素分子印迹材料的吸附动力学曲线用于测定分子印迹聚合物对四环素的吸附速率。
四环素分子印迹材料的吸附选择性用于验证分子印迹聚合物对模板分子是否有特异性吸附作用。
四环素分子印迹材料的回收率是反应待测物在样品分析过程中损失的程度,损失越少,回收率越高,回收率高说明该分子印迹聚合物对模板分子四环素的选择性吸附越高。
本发明的四环素分子印迹材料,肉眼观察为白色粉末状,在扫描电子显微镜下呈现微球状,粒径均匀,为1~2μm。
测定实施例的四环素分子印迹材料(分子印迹聚合物)对四环素的吸附等温曲线,吸附量随四环素浓度的增加而增加。在色谱条件:c18反相色谱柱,流动相:v/v为1:4甲醇-乙腈a和0.03mol/l草酸b条件下,在牛奶样品中加入四环素、土霉素和金霉素混合标样,经过高性能液体色谱hplc检测,分子印迹聚合物对四环素、土霉素和金霉素的回收率为98.3%、51.5%和46.2%,显然对四环素的回收率均高于土霉素和金霉素的加标回收率,说明本发明方法得到的m-mips含有与四环素空间结构匹配,官能团互补的空穴,在m-mips中形成了能够识别四环素的特异性结合位点,因此对四环素具有特异性吸附作用,能有效提高四环素检测的准确度。通过测定四环素及其结构类似物土霉素和金霉素在分子印迹聚合物上的结合量,其平衡吸附量分别为91.25mg/g、33.84mg/g、20.84mg/g,显然对四环素的吸附量最大,显示出m-mips对四环素显示出更强的亲和力,四环素分子印迹材料对四环素具有良好的吸附选择性,有效提高四环素的检测分析效率。
用本发明的四环素分子印迹材料测定四环素残留量时,先制备分子印迹固相萃取小柱,选择1ml乙腈作为上样溶液,2ml5%甲醇作为淋洗液,3ml0.1mol/lkoh和2ml0.1mol/l二甲基乙二醇丙烯酸酯edta溶液作为洗脱液。在样品牛奶中加入四环素,土霉素和金霉素混合标样,在色谱条件:c18反相色谱柱,流动相:甲醇-乙腈(1:4,v/v)(a)和0.03mol/l草酸(b)检测波长355nm,进样量20μl下,经hplc来检测分子印迹固相萃取柱对四环素的残留量。
实施例1
一、制备纳米fe3o4粒子
室温下,将6.50gfecl3·6h2o溶于50%的乙醇中,fecl3·6h2o与乙醇质量比为:16:70,磁力搅拌,转速为200rmp,同时以2℃/min的升温速度,升温至40℃,持续搅拌下,加入0.40g盐酸羟胺,fecl3·6h2o与盐酸羟胺质量比为:16:1,搅拌反应5min后,用氨水调节ph至7,逐滴加入油酸8ml,5min后,再以3℃/min的升温速度,升温至60℃,搅拌30min,反应结束,自然冷却至室温,用磁铁将黑色沉淀分离出来,并用20℃的无水乙醇洗涤,真空干燥,得到纳米fe3o4颗粒。
二、制备fe3o4@tio2复合颗粒
按钛酸丁酯tbt与无水乙醇体积比为1:6,将钛酸丁酯在无水乙醇中超声分散得到tbt分散液,按钛酸丁酯与盐酸摩尔比为2:0.01,将盐酸加入到tbt分散液中,直接在50℃下催化水解2h,空气中自然冷却至25℃,在转速200r/min,水浴温度25℃下,按钛酸丁酯与甘油体积比为1:5,加入模板溶液甘油,封口,室温下静置凝胶15d,待凝胶干燥后真空干燥、研磨,然后用水作为溶剂索式抽提,除去模板溶液,再真空干燥,得到介孔tio2。
将1ml去离子水、12ml无水乙醇、0.1ml浓盐酸配成溶液待用,去离子水与无水乙醇体积比为1:12,无水乙醇与浓盐酸体积比为12:0.1,配制成去离子水、无水乙醇、浓盐酸溶液。
将8g介孔tio2在12ml无水乙醇中超声分散得到介孔tio2分散液,介孔tio2与无水乙醇体积比为2:3,按纳米fe3o4颗粒与介孔tio2质量比为0.4:8,将0.4g纳米fe3o4颗粒放入三口瓶中,加入介孔tio2分散液,在50℃,转速200r/min下,水浴搅拌反应2min,溶胶,在纳米fe3o4颗粒表面包覆介孔tio2。
将去离子水、无水乙醇、浓盐酸溶液用恒压滴液漏斗,压力为88.35kpa,速度控制在0.1ml/s,逐滴加入至三口瓶中,室温下反应2h,凝胶,将反应产物转移至烧杯中静置6h,将沉淀物按现有技术真空干燥,得到fe3o4@tio2复合颗粒。
三、制备fe3o4@tio2-mps复合粒子
将0.4gfe3o4@tio2复合颗粒超声分散于50ml无水乙醇中,再加入0.6mlmps,fe3o4@tio2复合颗粒与mps质量比2:3,加入0.5ml吡啶,在转速200r/min下,同时以3℃/min的升温速度,升温至30℃,机械搅拌反应24h后,用磁铁分离反应产物,将上清液与沉淀物分离,用无水乙醇洗涤3次、砂心漏斗抽滤、真空干燥后,得到fe3o4@tio2-mps复合粒子。
四、制备fe3o4@tio2-mps-tfmaa复合粒子
将1.0gfe3o4@tio2-mps复合粒子,加入120ml水中超声分散均匀后,置于可加热磁力搅拌器上,在搅拌转速为200r/min条件下,以3℃/min的升温速度,升温至70℃,持续搅拌下,按质量比fe3o4@tio2-mps复合粒子:tfmaa:(nh4)2s2o8为1:6:0.02,先加入6mltfmaa,充入氮气除氧20min,氮气流量为0.1l/min,再加入0.02g(nh4)2s2o8作为引发剂进行接枝聚合反应,接枝聚合反应24h后,在索式抽提器中用无水乙醇抽提24h,得到的固体物质,将固体物质在60℃、真空度为133pa条件下,干燥24h,得到fe3o4@tio2-mps-tfmaa复合粒子。
五、制备饱和吸附了四环素的复合粒子
将1.0gfe3o4@tio2-mps-tfmaa复合粒子于40ml四环素溶液中,四环素溶液浓度为10mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比为6:7,恒温振荡5h,振荡频率100r/min、功率为1680w,使fe3o4@tio2-mps-tfmaa复合粒子饱和吸附四环素,过滤出微粒,真空干燥,得到饱和吸附了四环素的复合粒子。
六、制备四环素分子印迹材料
将饱和吸附了四环素的复合粒子1.0g,于40ml四环素水溶液中,四环素水溶液浓度为4mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比为6:7,用naoh调节ph为7,按饱和吸附了四环素的复合粒子与egde质量比为1:2.2,加入2mlegde,磁力搅拌,转速为200r/min,同时以3℃/min的升温速度,升温至50℃,反应8h,反应结束后用水和甲醇在砂芯漏斗中抽滤、清洗至中性,在索氏提取器中用体积比为9:1的甲醇与冰乙酸混合溶液抽提24h,除去模板分子四环素和未反应的egde,得到聚合物,然后用甲醇作为溶剂索式抽提,除去聚合物表面的乙酸,真空干燥,得到四环素分子印迹材料m-mips。
采用实施例1得到的四环素分子印迹材料,做吸附等温线实验得到四环素分子印迹材料对四环素的最大吸附量为89.5mg/g。做吸附动力学曲线显示7min达到吸附平衡。吸附选择性实验通过测定四环素及其结构类似物土霉素和金霉素在分子印迹聚合物上的结合量其平衡吸附量分别为89.5mg/g、30.6mg/g、23.3mg/g。四环素分子印迹材料对四环素、土霉素、金霉素的加标回收率为88.1%、49.3%、40.1%。
实施例2
一、制备纳米fe3o4粒子
室温下,将6.76gfecl3·6h2o溶于50%的乙醇中,fecl3·6h2o与乙醇质量比为:17:75,磁力搅拌,转速为300rmp,同时以3℃/min的升温速度,升温至50℃,持续搅拌下,加入0.42g盐酸羟胺,fecl3·6h2o与盐酸羟胺质量比为17:1,搅拌反应5min后,用氨水调节ph至9,逐滴加入油酸8.3ml,10min后,再以4℃/min的升温速度,升温至70℃,搅拌30min,反应结束,自然冷却至室温,用磁铁将黑色沉淀分离出来,并用20℃的无水乙醇洗涤,真空干燥,得到纳米fe3o4颗粒。
如图2所示,fe3o4颗粒呈球形,易发生团聚,平均粒径不超过25nm。
二、制备fe3o4@tio2复合颗粒
按钛酸丁酯tbt与无水乙醇体积比为1:7,将钛酸丁酯在无水乙醇中超声分散得到tbt分散液,按钛酸丁酯与盐酸摩尔比为1:0.01,将盐酸加入到tbt分散液中,直接在60℃下催化水解3h,空气中自然冷却至25℃,在转速300r/min,水浴温度25℃下,按钛酸丁酯与甘油体积比为1:8,加入模板溶液甘油,封口,室温下静置凝胶18d,待凝胶干燥后真空干燥、研磨,然后用水作为溶剂索式抽提,除去模板溶液,再真空干燥,得到介孔tio2。
将3ml去离子水、36ml无水乙醇、0.2ml浓盐酸配成溶液待用,去离子水与无水乙醇体积比为1:12,无水乙醇与浓盐酸体积比为18:0.1,配制成去离子水、无水乙醇、浓盐酸溶液。
将9g介孔tio2在36ml无水乙醇中超声分散得到介孔tio2分散液,介孔tio2与无水乙醇体积比为1:4,按纳米fe3o4颗粒与介孔tio2质量比为0.5:9,将0.5g纳米fe3o4颗粒放入三口瓶中,加入介孔tio2分散液,在40℃,转速300r/min下,水浴搅拌反应2min,溶胶,在纳米fe3o4颗粒表面包覆介孔tio2。
将去离子水、无水乙醇、浓盐酸溶液用恒压滴液漏斗,压力为88.35kpa,速度控制在0.1ml/s,逐滴加入至三口瓶中,室温下反应2h,凝胶,将反应产物转移至烧杯中静置10h,将沉淀物按现有技术真空干燥,得到fe3o4@tio2复合颗粒。
三、制备fe3o4@tio2-mps复合粒子
将0.5gfe3o4@tio2复合颗粒超声分散于50ml无水乙醇中,再加入0.625mlmps,fe3o4@tio2复合颗粒与mps质量比1:1.3,加入1.0ml吡啶,在转速300r/min下,同时以4℃/min的升温速度,升温至50℃,机械搅拌反应24h后,用磁铁分离反应产物,将上清液与沉淀物分离,用无水乙醇洗涤3次、砂心漏斗抽滤、真空干燥后,得到fe3o4@tio2-mps复合粒子。
四、制备fe3o4@tio2-mps-tfmaa复合粒子
将2.0gfe3o4@tio2-mps复合粒子,加入150ml水中超声分散均匀后,置于可加热磁力搅拌器上,在搅拌转速为300r/min条件下,以4℃/min的升温速度,升温至70℃,持续搅拌下,按质量比fe3o4@tio2-mps复合粒子:tfmaa:(nh4)2s2o8为1:5:0.021,先加入7mltfmaa,充入氮气除氧30min,氮气流量为0.1l/min,再加入0.042g(nh4)2s2o8作为引发剂进行接枝聚合反应,接枝聚合反应24h后,在索式抽提器中用无水乙醇抽提24h,得到的固体物质,将固体物质在60℃、真空度为133pa条件下,干燥24h,得到fe3o4@tio2-mps-tfmaa复合粒子。
五、制备饱和吸附了四环素的复合粒子
将2.0gfe3o4@tio2-mps-tfmaa复合粒子于50ml四环素溶液中,四环素溶液浓度为10mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比为6:7,恒温振荡6h,振荡频率100r/min、功率为1680w,使fe3o4@tio2-mps-tfmaa复合粒子饱和吸附四环素,过滤出微粒,真空干燥,得到饱和吸附了四环素的复合粒子。
六、制备四环素分子印迹材料
将饱和吸附了四环素的复合粒子2.0g,于50ml四环素水溶液中,四环素水溶液浓度为4mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比为6:7,用naoh调节ph为8,按饱和吸附了四环素的复合粒子与egde质量比为1:1.4,加入2.5mlegde,磁力搅拌,转速为300r/min,同时以4℃/min的升温速度,升温至50℃,反应8h,反应结束后用水和甲醇在砂芯漏斗中抽滤、清洗至中性,在索氏提取器中用体积比为9:1的甲醇与冰乙酸混合溶液抽提24h,除去模板分子四环素和未反应的egde,得到聚合物,然后用甲醇作为溶剂索式抽提,除去聚合物表面的乙酸,真空干燥,得到四环素分子印迹材料m-mips。
如图3所示,实施例2的四环素分子印迹材料呈现微球状,颗粒分布均匀,粒径为1~2μm,fe3o4@tio2-mps表面均匀地包覆有印迹层。
如图4所示,实施例2的四环素分子印迹材料,吸附等温线显示分子印迹材料对四环素的最大吸附量为91.2mg/g。
如图5所示,实施例2的四环素分子印迹材料,做吸附动力学曲线,在6min后吸附达到平衡。
如图6所示,实施例2的四环素分子印迹材料,吸附选择性表示通过测定四环素及其结构类似物土霉素和金霉素在分子印迹聚合物上的结合量,其平衡吸附量分别为91.2mg/g、33.8mg/g、20.8mg/g。
经高效液体色谱hplc检测,分子印迹材料对四环素、土霉素、金霉素的加标回收率为98.3%、51.5%、46.2%。
实施例3
一、制备纳米fe3o4粒子
室温下,将6.80gfecl3·6h2o溶于50%的乙醇中,fecl3·6h2o与乙醇质量比为18:80,磁力搅拌,转速为40rmp,同时以5℃/min的升温速度,升温至60℃,持续搅拌下,加入0.44g盐酸羟胺,fecl3·6h2o与盐酸羟胺质量比为16:1.1,搅拌反应5min后,用氨水调节ph至8,逐滴加入油酸9ml,15min后,再以6℃/min的升温速度,升温至80℃,搅拌30min,反应结束,自然冷却至室温,用磁铁将黑色沉淀分离出来,并用20℃的无水乙醇洗涤,真空干燥,得到纳米fe3o4颗粒。
二、制备fe3o4@tio2复合颗粒
按钛酸丁酯tbt与无水乙醇体积比为2:8,将钛酸丁酯在无水乙醇中超声分散得到tbt分散液,按钛酸丁酯与盐酸摩尔比为2:0.02,将盐酸加入到tbt分散液中,直接在70℃下催化水解4h,空气中自然冷却至25℃,在转速400r/min,水浴温度25℃下,按钛酸丁酯与甘油体积比为2:10,加入模板溶液甘油,封口,室温下静置凝胶20d,待凝胶干燥后真空干燥、研磨,然后用水作为溶剂索式抽提,除去模板溶液,再真空干燥,得到介孔tio2。
将2ml去离子水、24ml无水乙醇、0.2ml浓盐酸配成溶液待用,去离子水与无水乙醇体积比为1:12,无水乙醇与浓盐酸体积比为12:0.1,配制成去离子水、无水乙醇、浓盐酸溶液。
将10g介孔tio2在40ml无水乙醇中超声分散得到介孔tio2分散液,介孔tio2与无水乙醇体积比为1:4,按纳米fe3o4颗粒与介孔tio2质量比为0.3:10,将0.3g纳米fe3o4颗粒放入三口瓶中,加入介孔tio2分散液,在60℃,转速400r/min下,水浴搅拌反应3min,溶胶,在纳米fe3o4颗粒表面包覆介孔tio2。
将去离子水、无水乙醇、浓盐酸溶液用恒压滴液漏斗,压力为88.35kpa,速度控制在0.1ml/s,逐滴加入至三口瓶中,室温下反应2h,凝胶,将反应产物转移至烧杯中静置12h,将沉淀物按现有技术真空干燥,得到fe3o4@tio2复合颗粒。
三、制备fe3o4@tio2-mps复合粒子
将0.6gfe3o4@tio2复合颗粒超声分散于50ml无水乙醇中,再加入0.7mlmps,fe3o4@tio2复合颗粒与mps质量比1:1.2,加入1.0ml吡啶,在转速400r/min下,同时以6℃/min的升温速度,升温至40℃,机械搅拌反应24h后,用磁铁分离反应产物,将上清液与沉淀物分离,用无水乙醇洗涤3次、砂心漏斗抽滤、真空干燥后,得到fe3o4@tio2-mps复合粒子。
四、制备fe3o4@tio2-mps-tfmaa复合粒子
将3.0gfe3o4@tio2-mps复合粒子,加入180ml水中超声分散均匀后,置于可加热磁力搅拌器上,在搅拌转速为400r/min条件下,以6℃/min的升温速度,升温至70℃,持续搅拌下,按质量比fe3o4@tio2-mps复合粒子:tfmaa:(nh4)2s2o8为1.5:5.6:0.025,先加入8mltfmaa,充入氮气除氧30min,氮气流量为0.1l/min,再加入0.05g(nh4)2s2o8作为引发剂进行接枝聚合反应,接枝聚合反应24h后,在索式抽提器中用无水乙醇抽提24h,得到的固体物质,将固体物质在60℃、真空度为133pa条件下,干燥24h,得到fe3o4@tio2-mps-tfmaa复合粒子。
五、制备饱和吸附了四环素的复合粒子
将3.0gfe3o4@tio2-mps-tfmaa复合粒子于50ml四环素溶液中,四环素溶液浓度为10mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比为6:7,恒温振荡6h,振荡频率100r/min、功率为1680w,使fe3o4@tio2-mps-tfmaa复合粒子饱和吸附四环素,过滤出微粒,真空干燥,得到饱和吸附了四环素的复合粒子。
六、制备四环素分子印迹材料
将饱和吸附了四环素的复合粒子3.0g,于50ml四环素水溶液中,四环素水溶液浓度为4mmol/l,用50%的甲醇与乙腈溶液配置,甲醇与乙腈体积比为6:7,用naoh调节ph为8,按饱和吸附了四环素的复合粒子与egde质量比为1:1.1,加入3mlegde,磁力搅拌,转速为400r/min,同时以6℃/min的升温速度,升温至50℃,反应8h,反应结束后用水和甲醇在砂芯漏斗中抽滤、清洗至中性,在索氏提取器中用体积比为9:1的甲醇与冰乙酸混合溶液抽提24h,除去模板分子四环素和未反应的egde,得到聚合物,然后用甲醇作为溶剂索式抽提,除去聚合物表面的乙酸,真空干燥,得到四环素分子印迹材料m-mips。
实施例3得到的四环素分子印迹材料,吸附等温线显示四环素分子印迹材料对四环素的最大吸附量为86.2mg/g。做吸附动力学曲线得到在8min后吸附达到平衡。吸附选择性表示通过测定四环素及其结构类似物土霉素和金霉素在分子印迹聚合物上的结合量,其平衡吸附量分别为86.2mg/g、29.4mg/g、20.1mg/g。经hplc检测,四环素分子印迹材料对四环素、土霉素、金霉素的加标回收率为85.2%、46.4%、40.5%。
按照与文献1相同的方法制备出四环素印迹聚合物,作为对比例1,经测定其对四环素的最大吸附量为42mg/g;经8小时后吸附达到平衡;在牛奶样品中加入四环素,经hplc检测,印迹聚合物对四环素的加标回收率为80.3%。
按照与文献2相同的方法制备出四环素分子印迹整体柱,作为对比例2,经测定其对四环素的最大吸附量为55mg/g;经6小时后达到吸附平衡;经hplc检测,印迹聚合物对四环素在牛奶样品中的加标回收率为89.6%。
吸附量实验:分别称取0.05g实施例1、实施例2、实施例3的m-mips,将3份于锥形瓶中,分别加入浓度为1、2、3mmol/l的四环素甲醇-乙腈(6:7,v/v)标准溶液10ml,在室温下振荡吸附20h,4000转下离心5min,取上清液,过0.45μm膜,在355nm下用紫外分光光度计测定吸附后上清液中四环素的吸光度,根据公式(1)计算四环素表面分子印迹聚合物对四环素tc的吸附量。
式(1)中,q为吸附量mg/g,c0和ce分别为四环素初始浓度和吸附平衡时溶液中四环素浓度mg/l,v为溶液体积ml,m为实施例1、实施例2、实施例3中m-mips的质量g。其中,实施例2m-mips对四环素的吸附等温线如图4所示。实施例1、实施例2、实施例3、对比例1、对比例2的最大吸附量如表1。
表1实施例1、2、3的m-mips,对比例1的四环素印迹聚合物,2的四环素分子印迹整体柱,对四环素的最大吸附量
从图4可以看出,实施例2的四环素分子印迹材料对四环素的吸附量随四环素浓度的增加而增大,当四环素浓度为1.5mmol/l时,四环素分子印迹材料的吸附量为91.2mg/g,趋于平衡;相比对比例1的四环素印迹聚合物,实施例2的四环素分子印迹材料对四环素的吸附量最大,说明实施例2与对比例1四环素印迹聚合物的结构不同,所体现的对外吸附四环素的特性也不同。实施例2的四环素分子印迹材料含有与四环素空间结构匹配,官能团互补的空穴,在四环素分子印迹材料中形成有能够有效识别四环素的特异性结合位点,因此对四环素具有更强的吸附作用。
从表1可以看出,当四环素浓度为1.5mmol/l时,实施例2的四环素分子印迹材料对四环素的吸附量最大,为91.2mg/g,之后趋于平衡。
吸附动力学实验:分别称取0.05g实施例1、实施例2、实施例3的m-mips于25ml锥形瓶中,分别加入10ml、8mmol/l的四环素溶液,室温25℃下振荡,每隔半小时取出上清液,离心过0.45μm滤膜,在355nm下测定吸附后溶液的吸光度,按式(1)计算m-mips在不同时间内对四环素的吸附量。其中实施例2m-mips对四环素的吸附动力学曲线如图5所示。
从图5可以看出,实施例2的四环素分子印迹材料m-mips在0.5~6min内,m-mips对四环素的吸附量呈明显的上升趋势,在6min之后,吸附达到平衡。m-mips对tc的吸附主要有两种方式,一种是特异性吸附,另一种是非特异性吸附。吸附初期以特异性吸附为主,m-mips表面存在大量的印迹位点,所以四环素容易结合到印迹位点上,随着印迹位点被占用,吸附速率会显著减缓,直至吸附过程达到平衡状态。实施例2的四环素分子印迹材料m-mips的吸附速度很快,6min就达到吸附平衡,说明m-mips表面存在大量能特异性识别四环素的印迹空穴。
吸附选择性实验:准确称取0.02g实施例1、实施例2、实施例3的m-mips于6组锥形瓶中,分别加入2mmol/l四环素及其结构类似物土霉素、金霉素的甲醇溶液于6组锥形瓶中。根据吸附前后溶液中浓度变化,利用公式(1)计算得到实施例1、实施例2、实施例3的m-mips对不同物质的平衡吸附量q。其中实施例2的m-mips对四环素及其结构类似物土霉素、金霉素的吸附选择性如图6所示,可以看出,实施例2的m-mips对结构类似物土霉素和金霉素的吸附量没有明显差别,而对模板分子四环素却有较大差别。在结构组成上,四环素、土霉素、金霉素有相似的官能团,但是实施例2的m-mips却对四环素的吸附量最大,显示出更强的亲和力。分子印迹聚合物的识别过程受空穴大小、形状以及活性官能团位置共同作用,在印迹聚合物表面既存在与四环素结合的功能基团,又存在与四环素的空间结构互补、特定形状的空穴,这两个条件决定了印迹聚合物m-mips对模板分子的选择结合特性,可以达到很好的选择性富集效果。
实际样品的测定:对实施例1、实施例2、实施例3的m-mips、对比例1的四环素印迹聚合物、对比例2的四环素分子印迹整体柱,分别制备固相萃取小柱,选择1ml乙腈作为上样溶液,2ml5%甲醇作为淋洗液,3ml0.1mol/lkoh和2ml0.1mol/l二甲基乙二醇丙烯酸酯edta溶液作为洗脱液。在色谱条件:c18反相色谱柱,流动相:甲醇-乙腈(1:4,v/v)(a)和0.03mol/l草酸(b),检测波长355nm,进样量20μl下,牛奶样品中加入四环素,土霉素和金霉素混合标样,加标量为以下三个浓度:300μg·l-1,500μg·l-1和700μg·l-1,经过hplc检测,得到牛奶样品经实施例2m-mips固相萃取柱处理后的色谱图如图7所示;分子印迹固相萃取柱对四环素,土霉素和金霉素的回收率和相对标准偏差rsd结果如表2。
表2实施例1、实施例2、实施例3的m-mips、对比例1的四环素印迹聚合物、对比例2的四环素分子印迹整体柱在牛奶样品中对四环素、土霉素和金霉素加标回收率
从表2可以看出,实施例1、实施例2、实施例3的m-mips、对比例1的四环素印迹聚合物、对比例2的四环素分子印迹整体柱对四环素的回收率均高于土霉素和金霉素的加标回收率,实施例2的四环素分子印迹材料对四环素的回收率均高于对比例1的四环素印迹聚合物、对比例2的四环素分子印迹整体柱的回收率。表明实施例2的四环素分子印迹材料对四环素的选择性更高。
如图7所示,a为牛奶样品中加入四环素、土霉素和金霉素后的标准样品的色谱图,b为标准样品经实施例2m-mips萃取后的色谱图。从图7中可以看出用实施例2m-mips处理样品后得到四环素的含量比标准样品的更高。这再一次证明实施例2的四环素分子印迹材料具备特异性识别四环素的能力。
通过对比可以看出,本发明的四环素表面分子印迹聚合物材料,磁性强,吸附速度快,效率高,识别性能好,选择性高,比表面积大,吸附容量大,能够快速识别并分离食品样品中的四环素。
- 还没有人留言评论。精彩留言会获得点赞!