一种氟喹诺酮类中间体2,4-二氯-5-氟苯甲酰氯的制备方法与流程
本发明涉及一种氟喹诺酮类中间体的制备方法,特别是涉及一种氟喹诺酮类中间体2,4-二氯-5-氟苯甲酰氯的制备方法,属于药物合成技术领域。
背景技术:
喹诺酮类是人工合成的含4-喹诺酮母核为基本结构的抗菌药物,从20世纪70年代后期诺氟沙星问世以来,第三代喹诺酮类-氟喹诺酮类药物的研究和开发引起了抗菌药物的革命,出现了很多有临床价值的新药,如氧氟沙星、环丙沙星、罗美沙星、氟罗沙星等,成为临床主要的抗感染药物之一,仅次于头孢菌类和青霉素类药物。氟喹诺酮类药物的结构特点是在6-位有氟原子,在7-位有取代氨基,文献报道的合成路线很多,但这些原料来源困难,且价格高。
大部分是从氟氯苯乙酮(2,4-二氯-5-氟苯乙酮)为起始原料,与碳酸二乙酯缩合,再与原甲酸三乙酯进行乙氧亚甲基化,与不同胺胺化,最后经环合和不同的哌嗪取代完成,该路线主要缺点是路线中两步使用了活性高的钠氢,存在安全隐患,合成路线如下:
目前用2,4-二氯氟苯作为原料,制备2,4-二氯-5-氟苯甲酰氯中间体,与不同的胺基丙烯酸甲酯缩合,经环合、取代得到氟喹诺酮类,此合成路线较短,安全性高,操作简单,合成路线如下:
随着沙星类药物需求量不断夸大,其合成中的中间体2,4-二氯-5-氟苯甲酰氯的需求量也日益增加,关于它的合成方法也很多,安永彬等人以2,4-二氯氟苯等为原料,以三氯化铝为催化剂,naclo溶液为氧化剂,在70-80℃反应,总收率为78.4%,该方法使用次氯酸钠等高污染、高风险的原料,且次氯酸钠需大大过量,污染大、成本高(中国抗生素杂质29(2004):529-530),合成路线如下;
温新民等人提出了使用2,4-二氯氟苯与乙酰氯在三氯化铝的存在下反应制备2,4-二氯-5-氟苯乙酮,然后在硝酸作用氧化成酸,经过酰化的2,4-二氯-5-氟苯甲酰氯,该制备方法碱解反应需要过来的硝酸,造成成本的浪费,且酰化反应需要绝对无水,不适合放大生产(济宁医学院学报23(2000):21-22),合成路线如下:
selsaku.k等人使用2,4-二氯氟苯在alcl3催化下与四氯化碳反应引入三氯甲基,经酸性水解得到2,4-二氯-5-氟苯甲酸,再与二氯亚砜酰化得到2,4-二氯-5-氟苯甲酰氯。该制备方法第一步有约20%-30%的副产物(fcl2c6h2)2ccl2,使得原料的利用低,造成成本高,难以实现工业化生产(us5241111),合成路线如下:
为了降低2,4-二氯氟苯与四氯化碳反应时产生二聚物的比例,吴政杰等人发明了固体酸催化剂s2o82-/sm2o3-zro2-al2o3以及李乙刚等人发明了复合固体超强酸催化剂,在这些催化剂的作用下,生成2,4-二氯-5-氟-(三氯甲基)苯,再经fecl3催化水解即得。该方法虽然降低了二聚物的比例,但无法避免二聚物的生成;并且合成催化剂步骤繁琐,成本高,且活化温度高达到600℃,存在安全隐患,不适合放大生产(cn104725221、cn104649890),合成路线如下:
技术实现要素:
本发明的主要目的是解决现有技术中制备化合物2,4-二氯-5-氟苯甲酰氯存在安全隐患、成本较高及不利于工业化生产的问题,而提供一种氟喹诺酮类中间体2,4-二氯-5-氟苯甲酰氯的制备方法。
本发明的目的可以通过采用如下技术方案达到:
一种氟喹诺酮类中间体2,4-二氯-5-氟苯甲酰氯的制备方法,其反应方程式如下:
通过碱解反应将已制备的化合物iv转换成化合物v*及2,4-二氯氟苯,再通过酰化反应制备得到化合物2,4-二氯-5-氟苯甲酰氯。
化合物iv结构式如下:
化合物v*结构式如下:
该氟喹诺酮类中间体2,4-二氯-5-氟苯甲酰氯的制备方法,包括如下步骤:
步骤1:碱解反应
将制备好的化合物(iv)加热成熔融状态,加入碱性试剂,保持熔融状态,搅拌反应,加入溶剂,回流,充分反应得到化合物v*与2,4-二氯氟苯;
步骤2:酰化反应
控制温度在49-51℃,向化合物v*中慢慢滴加二氯化砜,升温至69-71℃,搅拌反应得化合物2,4-二氯-5-氟苯甲酰氯。
化合物iv与碱性试剂的摩尔比为1:1.5-2.5。
碱性试剂为naoh、koh、甲醇钠和乙醇钠中的至少一种。
溶剂为1,4-二氧六环和甲苯中的至少一种。
本发明的有益技术效果:
本发明提供的一种氟喹诺酮类中间体2,4-二氯-5-氟苯甲酰氯的制备方法,本发明通过将化合物iv转化为化合物v*与原料2,4-二氯氟苯,避免使用双氧水等易制爆物料,化合物v*通过一步反应,转化为化合物2,4-二氯-5-氟苯甲酰氯。本发明生成的起始原料2.3-二氯氟苯可以回收利用,同时羧酸盐(v*)转化为酰氯(ii),不产生盐酸,可以降低污染,这比现有的技术更有优势,增加资源利用率,降低生产风险,降低生产成本,更符合工业化生产。
附图说明
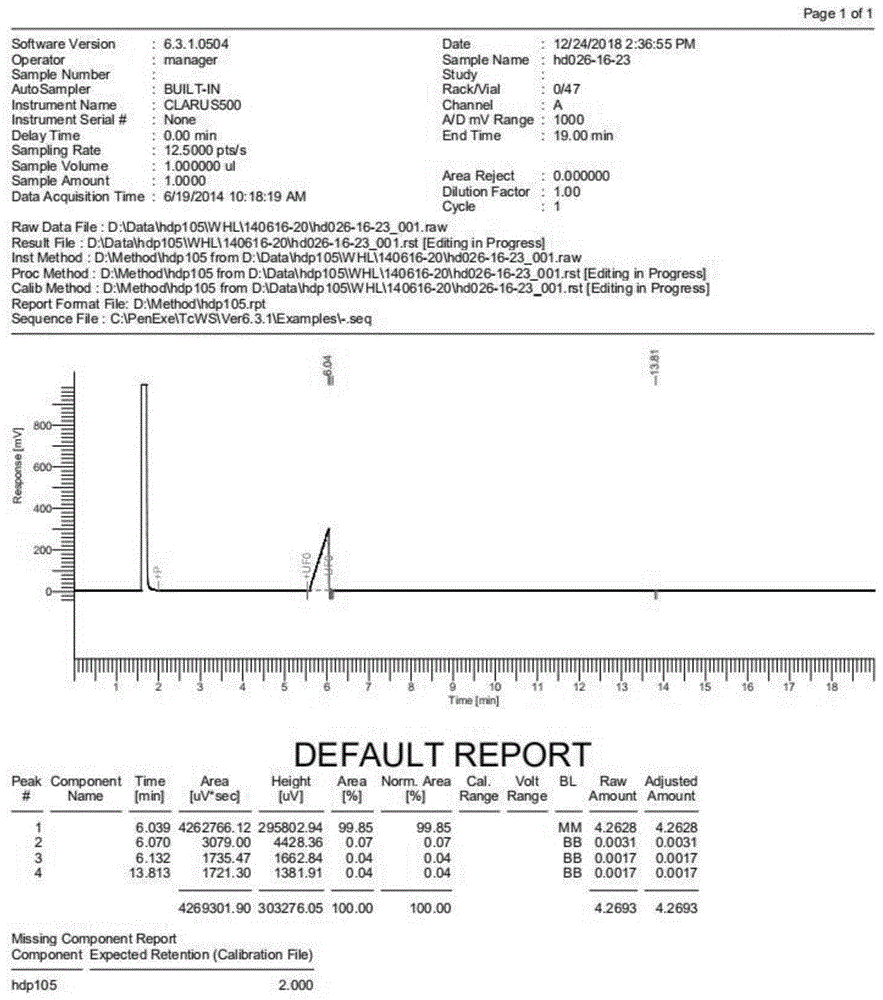
图1为化合物2,4-二氯氟苯的纯度图谱。
具体实施方式
为使本领域技术人员更加清楚和明确本发明的技术方案,下面对本发明作进一步详细的描述,但本发明的实施方式不限于此。
在本实施例中,本实施例提供的氟喹诺酮类中间体2,4-二氯-5-氟苯甲酰氯的制备方法,其反应方程式如下:
其是通过碱解反应将已制备的化合物iv转换成化合物v*及2,4-二氯氟苯,再通过酰化反应制备得到化合物2,4-二氯-5-氟苯甲酰氯。
化合物iv结构式如下:
化合物v*结构式如下:
该氟喹诺酮类中间体2,4-二氯-5-氟苯甲酰氯的制备方法,包括如下步骤:
步骤1:碱解反应
将制备好的化合物(iv)加热成熔融状态,加入碱性试剂,保持熔融状态,搅拌反应,加入溶剂,回流,充分反应得到化合物v*与2,4-二氯氟苯;
步骤2:酰化反应
控制温度在49-51℃,向化合物v*中慢慢滴加二氯化砜,升温至69-71℃,搅拌反应得化合物2,4-二氯-5-氟苯甲酰氯。
在本实施例中,本实施例提供将化合物2,4-二氯氟苯转化为化合物ii的方法,包括如下步骤:
步骤1:将原料2,4-二氯氟苯(100g,0.606mol)加入到四氯化碳(107.4g,0.667mol)中,搅拌溶解后,加入三氯化铁(1.52g,9.3mmol),升温至70℃并维持2小时,液相检测原料基本消失,降至室温,加入2.5mhclaq(500ml)淬灭反应,搅拌至澄清后分层,取下层有机相减压蒸馏,得化合物ⅰ(128.5g,75.1%)和釜残即化合物ⅲ(25.3g,20.3%);
步骤2:将无水三氯化铁(1g)加入到化合物ⅰ(20g,70.9mmol)中,加热至145℃后,缓慢滴加水(1.27g,70.9mmol),加完继续搅拌30分钟,气相检测原料消失,减压蒸馏得化合物ⅱ(15.8g,98.1%)。
其合成路线如下所示:
在本实施例中,为了更清楚地表达本发明的技术方案,本发明明提供如下实施例:
实施例1:
在反应瓶中加入化合物iv(100g),保持温度在90至100℃,化合物iv在熔融状态,加入氢氧化钠(1.5eq,16g),保持温度在90-100℃的条件下,搅拌1小时,让氢氧化钠充分分散开,慢慢升温到110℃左右,体系自行反应放热,并升温到140-150℃,水浴控制温度在120-130℃,此时hplc检测原料剩余10-30%,降温至110℃左右,一次性加入1,4-二氧六环(100ml),回流反应3h左右,hplc检测原料剩余小于1%,常压蒸馏,去除二氧六环与水,得到混合物化合物v*与2,4-二氯氟苯;
在上述混合物(化合物v*与2,4-二氯氟苯)中,保持温度在50℃左右,慢慢滴加二氯亚砜(1.3eq,42g),升温至70℃-80℃,搅拌反应2-3小时,反应结束后,减压去掉少量多余的二氯化砜,降压精馏,外温150-160℃收集2,4-二氯氟苯(40g),gc检测纯度大于99.9%,温度68-70℃,收集产品化合物2,4-二氯-5-氟苯甲酰氯(55g),gc纯度大于99.5%,2,4-二氯氟苯的回收率为90%,这2步的总收率为90%。
实施例2:
在反应瓶中加入化合物iv(500g),保持温度在90-100℃,化合物iv在熔融状态,加入氢氧化钠(1.5eq,80.4g),保持温度在90-100℃条件下,搅拌1小时,让氢氧化钠充分分散开,慢慢升温到110℃左右,体系自行反应放热,并升温到140-150℃,水浴控制温度在120-130℃,此时hplc检测原料剩余10-30%,降温至110℃左右,一次性加入1,4-二氧六环(500ml),回流反应3h左右,hplc检测原料剩余小于1%,常压蒸馏,去除二氧六环与水,得到混合物化合物v*与2,4-二氯氟苯;
在上述混合物(化合物v*与2,4-二氯氟苯)中,保持温度在50℃左右,慢慢滴加二氯亚砜(1.3eq,207g),升温至70℃-80℃,搅拌反应2-3小时,反应结束后,减压去掉少量多余的二氯化砜,降压精馏,外温150-160℃收集80%的2,4-二氯氟苯(199g),gc检测纯度大于99.9%,温度68-70℃,收集产品化合物2,4-二氯-5-氟苯甲酰氯(277g),gc纯度大于99.5%,2,4-二氯氟苯的回收率为90%,这2步的总收率为90%。
实施例3:
在反应瓶中加入化合物iv(800g),保持温度在90-100℃,化合物iv在熔融状态,加入氢氧化钠(1.5eq,129g),保持温度在90-100℃条件下,搅拌1小时,让氢氧化钠充分分散开,慢慢升温到110℃左右,体系自行反应放热,并升温到140-150℃,水浴控制温度在120-130℃,此时hplc检测原料剩余10-30%,降温至110℃左右,一次性加入1,4-二氧六环(800ml),回流反应3h左右,hplc检测原料剩余小于1%,常压蒸馏,去除二氧六环与水,得到混合物化合物v*与2,4-二氯氟苯;
在上述混合物(化合物v*与2,4-二氯氟苯)中,保持温度在50℃左右,慢慢滴加二氯亚砜(1.3eq,333g),升温至70℃-80℃,搅拌反应2-3小时,反应结束后,减压去掉少量多余的二氯化砜,降压精馏,外温150-160℃收集80%的2,4-二氯氟苯(319g),gc检测纯度大于99%,温度68-70℃,收集产品化合物2,4-二氯-5-氟苯甲酰氯(445g),gc纯度大于99.5%,2,4-二氯氟苯的回收率为90%,这2步的总收率为90%。
在上述实施例中,化合物ⅴ*的1hnmr谱图如下所示:
1hnmr(dmso300mhz):δ7.831-7.862(d,1h),7.921-7.943(d,1h).
在上述实施例中,化合物ii的1hnmr谱图如下所示:
1hnmr(cdcl3300mhz):δ7.600-7.736(d,1h),7.688-7.957(d,1h).
在上述实施例中,化合物2,4-二氯氟苯的纯度图谱如图1所示。
在上述实施例中,通过将化合物iv转化为化合物v*与原料2,4-二氯氟苯,避免使用双氧水等易制爆物料,化合物v*通过一步反应,转化为化合物2,4-二氯-5-氟苯甲酰氯,产品纯度较高,两步反应总收率较高,生成的起始原料2.3-二氯氟苯可以回收利用,同时羧酸盐(v*)转化为酰氯(ii),不产生盐酸,可以降低污染,这比现有的技术更有优势,增加资源利用率,降低生产风险,降低生产成本,更符合工业化生产。
以上所述,仅为本发明进一步的实施例,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明所公开的范围内,根据本发明的技术方案及其构思加以等同替换或改变,都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!